Daly Lab research
Research activities | Collaborations | Student research projects | Publications
About Professor Roger Daly
Professor Roger Daly was awarded his PhD from the University of Liverpool, UK before taking up positions as a postdoctoral fellow at the Imperial Cancer Research Fund, London, UK, followed by New York University Medical Centre, New York, USA. Roger established the Signal Transduction Group at the Garvan Institute of Medical Research in 1993, and relocated his laboratory to Monash University in March 2013, where he was Head of the Department of Biochemistry and Molecular Biology until April 2023 and current Co-Head of the Biomedicine Discovery Institute Cancer Program and Program Lead, Discovery and Innovation, Monash Partners Comprehensive Cancer Consortium (2023-). Professor Daly is an internationally recognised expert in growth factor receptor signalling and its deregulation in cancer. Over the last decade he has established cutting-edge technology platforms in mass spectrometry-based proteomics and kinomics. These have been successfully applied to the characterisation of cancer signalling networks with the goal of identifying novel therapeutic targets and biomarkers.
Our research
Current projects
- Phosphorylation-based signalling networks in cancer
- Intercellular communication in the tumour microenvironment
- Network-level interrogation of the PEAK family of pseudokinase scaffolds
Visit Professor Daly’s Monash research profile to see a full listing of current projects.
Research activities
Research vision:
Perturbations in cellular signalling play a fundamental role in human cancer and provide the rationale for many targeted therapies. However, it is becoming increasingly evident that network-level insights are required to fully explain the impact of oncogenic perturbations, or resistance to specific therapies. Furthermore, integrated application of multiple ‘omic approaches with computational biology will be essential if we are to characterize such networks, understand their dynamic behaviour and fully exploit their therapeutic potential.
Over the last decade my laboratory has applied cutting-edge technology platforms in mass spectrometry (MS)-based proteomics to define key oncogenic signalling networks, placing my research at the forefront of the field. My research vision is to exploit this competitive position and undertake systems-level interrogation of specific intra- and intercellular signalling networks in order to identify novel therapeutic targets and biomarkers for triple negative breast cancer (TNBC), high-risk, localized prostate cancer (PC) and pancreatic ductal adenocarcinoma (PDAC). This will be achieved via an integrated approach coupled to a powerful research translation pipeline (Figure 1).

Figure 1. Schematic of the Signalling Network Laboratory’s overall research program to identify novel therapeutic targets and biomarkers. PDX: patient derived xenograft.
1. Phosphorylation-based signalling networks in cancer
Protein phosphorylation is one of the most important and central regulatory mechanisms of cell signalling pathways. The dynamic and reversible phosphorylation of proteins, regulated by kinases and phosphatases under the control of complex signalling networks, modulates protein activity, binding interactions and subcellular localisation that in turn impacts upon diverse biological processes such as cell proliferation, apoptosis, migration and metabolism. Due to the importance of phosphorylation in cellular signalling networks it is not surprising that perturbations result in numerous diseases including cancer. Our laboratory has established cutting edge, mass spectrometry (MS)-based techniques that enable us to detect and quantify cellular phosphorylation events in a global fashion, providing novel insights into oncogenic signalling networks and how they differ between particular cancers and cancer subtypes.
1a. Interrogation of the SFK signalling network in TNBC
Our recent characterization of the Src-regulated kinome (Ma et al. Nat Comms, 2019) provided mechanistic insights into Src-induced transformation and also highlighted improved therapeutic strategies. Several of the kinases identified (Figure 2), however, are poorly characterized, particularly MAP3K6 and MAP4K5, and regulated by Src-regulated phosphorylation events of unknown function. We plan to address these issues via detailed mechanistic and functional characterization that will include delineation of the role of Src-induced phosphorylation, identification of kinase substrates by quantitative MS-based proteomics, determination of whether particular kinases modulate sensitivity to Src-directed therapy, and characterization of expression patterns in TNBC patient tissue cohorts. Temporal maps of kinase regulation by Src will be incorporated into computational models in collaboration with our collaborator Dr Lan Nguyen.
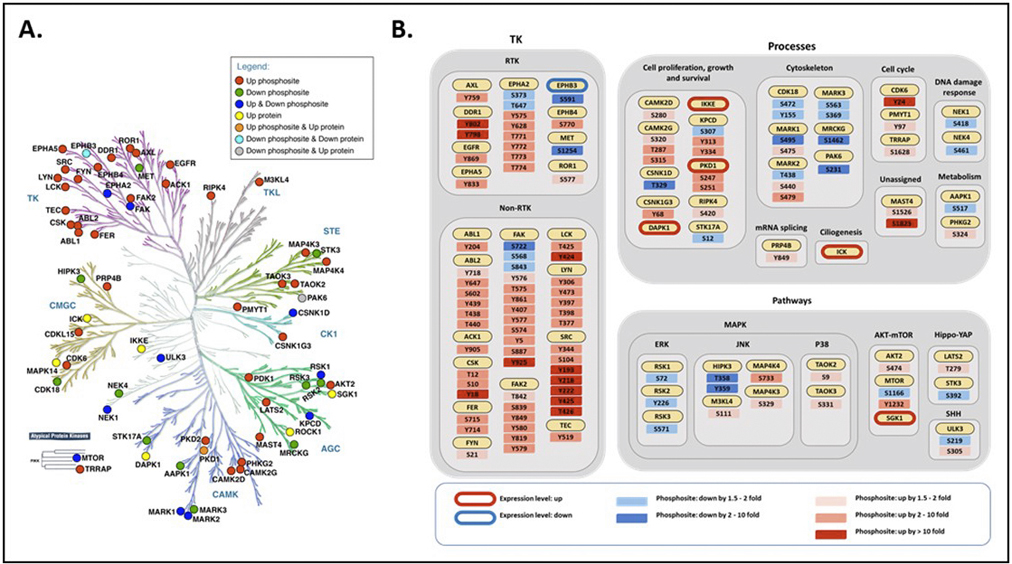
Figure 2. A. Distribution of Src-regulated kinases over the human kinome tree. Regulation at the expression and/or phosphorylation level is indicated by corresponding shading, as defined in the legend. B. Assignment of Src-regulated kinases to specific kinase classes, biological processes and pathways. Up or down means levels were increased or decreased in Src cells compared to control cells, respectively.
1b. Receptor tyrosine kinase (RTK) network re-modelling in TNBC
Whilst phosphoproteomic profiling of triple negative breast (TNBC) cell lines and patient derived xenografts (PDX) within the laboratory has identified numerous potential therapeutic targets, often the response of cells to target inhibition is limited by network remodelling. The latter effect will be characterized globally by MS-based phosphoproteomics, and how specific RTKs affect cellular response determined via pharmacological and siRNA-based approaches. In collaboration with Lan Nguyen, we are building a predictive computational model of specific RTK signalling networks in TNBC and will apply his Drug Combination Discovery Framework (DCDF), where the co-inhibition effects of all possible pair-wise combinations of network nodes are systematically evaluated, and compared, based on well-established quantitative drug synergy metrics (Figure 3). This provides a rational approach for ranking and prioritising drug combinations, thereby greatly enhancing the efficiency of therapeutic strategy development. Prioritized drug combinations will then be tested in appropriate in vitro, ex vivo and in vivo models, including TNBC organoids.

Figure 3. Schematic of computational modelling workflow
Outcomes: Identification of improved therapeutic strategies for TNBC through integrative analysis of RTK and SFK signalling networks
1c. Targeting pancreatic cancer by interrogation of tyrosine kinase signalling networks
The most common type of pancreatic cancer is pancreatic ductal adenocarcinoma (PDAC), making up 85 per cent of cases. Currently only five per cent of people with this type of cancer survive longer than five years after their diagnosis, and aggressive chemotherapy is currently the standard treatment. Consequently, there is an urgent need for improved, personalized treatment strategies.
Exploiting our expertise in mass spectrometry we characterized global tyrosine phosphorylation patterns across large panels of human PDAC cell lines (Humphrey et al MCP 2016). This provided a novel taxonomy for PDAC by identifying subtype-selective signalling networks. While previous studies indicated that pancreatic cancer could be subclassified based on gene expression, this was the first time subclassification had been achieved in this manner.
Of particular interest, one subtype was characterised as having enhanced signalling through various receptor tyrosine kinases (RTKs). Importantly, cell lines from this subtype showed enhanced sensitivity to the drug erlotinib, which is a small molecule inhibitor of a particular RTK and used for PDAC treatment. This work therefore provides 'proof-of-principle' that our approach can be used to identify predictive biomarkers for personalised patient treatment.
We are now expanding on these exciting findings to identify and characterise other subtype-selective phosphosignatures that represent novel biomarkers and/or therapeutic targets. We are also progressing from cell lines to patient-derived mouse models of pancreatic cancer to provide more clinically relevant information that can lead to patient clinical trials.
2. Intercellular communication in the tumour microenvironment
The tumour microenvironment (TME) plays a major role in cancer development and progression, and offers novel opportunities for research translation. For example, in prostate cancer (PC), cancer-associated fibroblasts (CAFs), unlike normal prostate fibroblasts (NPFs), promote tumourigenesis by BPH-1 prostate epithelial cells, and we have identified the LOXL2/collagen/DDR2 signalling axis as an important mediator of signalling between CAFs and prostate cancer cells (Figure 4). To interrogate signalling within the TME, we are exploiting a new approach, termed CTAP. This distinguishes the proteomes of two different cell types in co-culture without the need for their separation, which would disrupt intercellular signalling, and reveals how heterocellularity extends cellular capacity beyond canonical cell-autonomous pathways (Figure 5).

Figure 4. Workflow used to identify LOXL2 as a mediator of signalling between CAFs and prostate cancer cells.
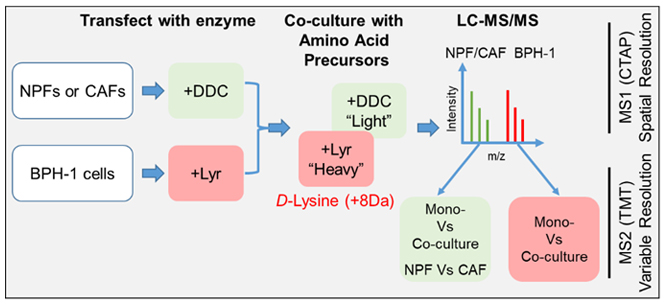
Figure 5. Application of CTAP methodology to our intercellular communication model. CTAP provides spatial resolution, identifying which cell type the MS-detected peptides are derived from. Discrimination between additional variables is achieved by differential isobaric tandem mass tag (TMT) labelling of peptides. BPH-1 cells: prostate epithelial cell line.
The functional role of specific intercellular signalling pathways will be characterized using in vitro models as well as in vivo tissue recombination and an orthotopic xenograft model that exhibits metastatic potential. In addition, candidate biomarkers of prostate cancer development and poor prognosis will be evaluated using unique patient tissue cohorts accessed through collaboration with our collaborator Prof Lisa Horvath (Garvan Institute/Lifehouse, Sydney).
Outcomes: Characterisation of novel mechanisms of intercellular signalling between prostate epithelial or PC cells and NPFs/CAFs; delineation of novel therapies that block PC development or spread; identification of potential companion biomarkers for such therapies.
3. Network-level interrogation of the PEAK family of pseudokinase scaffolds
The PEAK family contains SgK269/PEAK1, SgK223/PEAK2, and the recently-identified c19orf35/PEAK3 (Figure 6). My group has determined that PEAK1 represents a master regulator of EGFR signalling over time, defined oncogenic roles for PEAK1 and PEAK2 in TNBC and pancreatic cancer, respectively, and determined that these proteins homo- and hetero-dimerize to diversify signal output (Figure 7). However, the protein interaction networks mediated by different PEAK complexes are yet to be defined. In addition, PEAK3 is currently uncharacterized. By defining the role of specific PEAK complexes, we will provide fundamental insights into how these scaffolds mediate oncogenic signalling that can be exploited for development of novel therapeutic approaches.
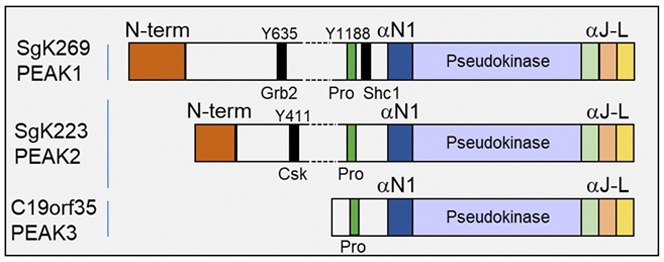
Figure 6. Schematic representation of PEAK family scaffolds. Pro: proline-rich region. αN1-aL: helical regions that mediate dimerization.
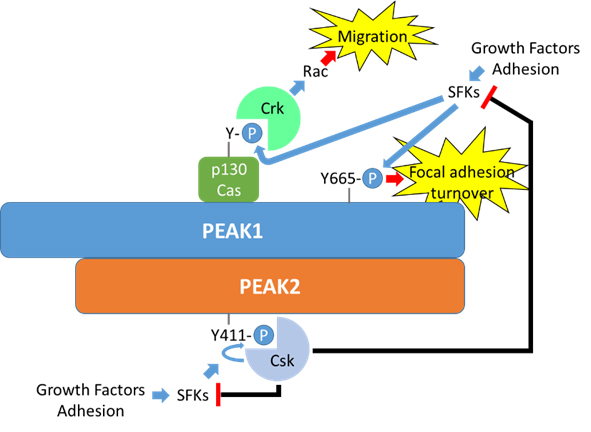
Figure 7. A potential role for PEAK1/PEAK2 heterotypic association in regulation of focal adhesion dynamics. Both PEAK1 and PEAK2 localize to focal adhesions, and PEAK1 is known to associate with specific focal adhesion components. PEAK1 also regulates focal adhesion turnover in a manner dependent on dynamic phosphorylation of Y665. Heterotypic association of PEAK1 with PEAK2, which recruits Csk, provides a potential mechanism for dynamic regulation of Src family kinases and hence focal adhesion turnover.
3a. Protein interaction networks mediated by specific scaffold complexes
Using BiCAP/MS methodology, which defines the interactome for specific bipartite complexes, we have identified interactors specific to the PEAK1 homodimer, PEAK2 homodimer, and the PEAK1/2 heterodimer. The same strategy will now be applied to the PEAK3 homodimer and PEAK1/3 and PEAK2/3 heterodimers. Data integration will define network maps for each of the 6 possible scaffold complexes, and associated pathways/processes will be defined using bioinformatics. Protein recruitment by these scaffolds in response to growth factor stimulation will be monitored, in parallel and over time, by scheduled multiple reaction monitoring (sMRM).
3b. Defining the functional role of specific scaffold complexes and their effectors
To determine the role of specific dimerization and oligomerization interfaces, wild-type and mutant PEAK family members will be reintroduced into CRISPR-generated knockout cell lines and key signalling and biological endpoints analysed. This work will define the functional role of specific PEAK1-3 homo- and heterotypic complexes and complement X-Ray crystallography and cryo-EM studies (collaboration with Isabelle Lucet, WEHI, Figure 8).
In addition, the mammary gland autonomous role of PEAK1 in tumour development and metastasis will be investigated using PEAK1 knockout mice (collaboration with Richard Klemke, UCSD).
Appropriate patient tissue cohorts will be also used to determine the relationship between expression of specific PEAK family proteins and their interactors and cancer evolution, clinicopathological features and patient outcome.
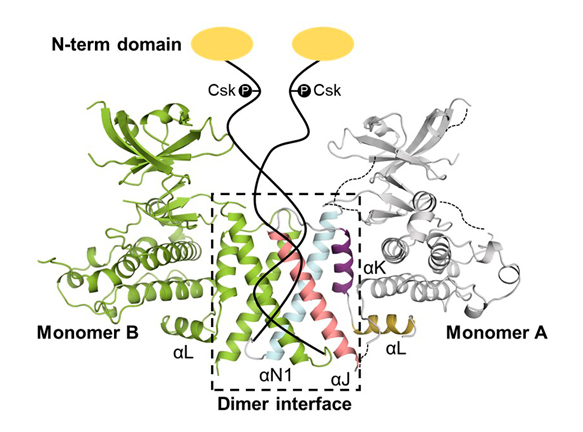
Figure 8. Structure of SgK223 pseudokinase domain and flanking helices, highlighting the dimerization interface (Patel et al. Nat Comms, 2017). The remainder of the protein is represented schematically.
Outcomes: Definition and functional annotation of protein interaction networks for specific PEAK complexes; identification of interaction interfaces and effectors that represent targets for drug discovery programs.
Techniques/expertise
- Cell Culture – 2D, 3D inc organoids
- Cellular imaging
- Mass Spectrometry - phosphoproteomics, kinomics, CTAP
- Western blot, IHC
- CRISPR
- sh/siRNA screens
Disease models
- Breast and gastric cancer organoids
- Knock-out mice
- Patient derived xenografts
- Orthotopic xenograft models
- In-vivo tissue recombination
Collaborations
We collaborate with many scientists and research organisations around the world. Some of our more significant national and international collaborators are listed below. Click on the map to see the details for each of these collaborators (dive into specific publications and outputs by clicking on the dots).
Cancer network biology:
J Wu (Peking University, China)
R Klemke (UCSD, USA)
C Jorgensen (CRUK Manchester, UK)
M Frame (Uni Edinburgh, UK)
E Fleuren (CCIA, Sydney)
I Lucet (WEHI, Melbourne)
L Nguyen (Monash)
J Song (Monash)
S Rosenbluh (Monash)
Y Khew-Goodall (Centre for Cancer Biology, Adelaide)
A Swarbrick (Garvan, Sydney)
C Ormandy (Garvan, Sydney)
M Pajic (Garvan, Sydney)
G Risbridger (Monash)
R Taylor (Monash)
C Mitchell (Monash)
K Simpson (VCFG/Peter MacCallum Cancer Centre, Melbourne)
Research Translation:
Breast cancer: Pathologists: S O’Toole (RPAH/Garvan - Sydney), E Millar (St George Hospital/Garvan - Sydney);
Medical oncologists: V Ganju (Monash Health), S Loi (Peter MacCallum Cancer Centre, Melbourne)
Prostate cancer: Pathologist: J Kench (RPAH/Garvan - Sydney)
Medical oncologist: L Horvath (RPAH/Garvan - Sydney)
Gastric cancer: Surgeon: X Shen (WMU, China)
Pancreatic cancer: Surgeon scientist: A Biankin (Uni Glasgow, UK) and national/international APGI/ICGC researchers
Student research projects
The Daly Lab offers a variety of Honours, Masters and PhD projects for students interested in joining our group. There are also a number of short term research opportunities available.
Please visit Supervisor Connect to explore the projects currently available in our Lab.