Ryan Lab research
Collaborations | Student research projects | Publications
About Professor Mike Ryan
Mike is Head of the Mitochondrial Biology and Disease Laboratory in the Monash Biomedicine Discovery Institute and Interim Deputy Vice-Chancellor (Research) and Senior Vice President at Monash University. Mike joined the Department of Biochemistry and Molecular Biology in 2014. Before this he was Head of the Department of Biochemistry at La Trobe University. After undertaking his PhD on molecular chaperones with Peter Hoj (La Trobe/Adelaide), Mike joined the group of Klaus Pfanner (University of Freiburg, Germany) as an Alexander von Humboldt fellow (1997-1999) where he studied protein import mechanisms. His group now uses a variety of techniques including gene editing, proteomics and mouse models to investigate and characterise the gene products involved in mitochondrial disease and dynamics.
Mike was President of the Australian Society for Biochemistry and Molecular Biology, a member of the Scientific and Medical Advisory Panel for the Australian Mitochondrial Disease Foundation (AMDF) and a member of the Australian Academy of Sciences National Committee for Biomedical Science. Mike was also on the Grants and Selection Panel and currently serves as Chair Panel of the International Human Frontier Science Program. In 2023, Mike was awarded the ASBMB Lemberg Medal for excellence in biochemistry and molecular biology and significant contributions to the field. His work is funded by the National Health and Medical Research Council, the Australian Research Council and the Medical Research Future Fund.
Our research
Current projects
- Gene editing and proteomic approaches to understand human mitochondrial protein function and disease
- Uncovering the link between the mitochondrial MICOS complex and programmed cell death
- Mitochondrial dynamics & neurodegeneration
Visit Professor Ryan's Monash research profile to see a full listing of current projects.
Research activities
Mitochondria are essential organelles and are present in all of our cells. They are known as the "powerhouse" of the cell since they burn carbon to make chemical energy (ATP). They are also the "poison-cupboard" of our cells since if the mitochondrial outer membrane is opened, proteins are released that cause cell death (apoptosis). Cell death is important since as our cells divide we must kill others - otherwise cancers may arise and so this process is very well regulated.
Mitochondria are also involved in aging, reactive oxygen species production and immune responses, and defects in mitochondria are implicated in many diseases including Parkinson's disease and Alzheimer's.
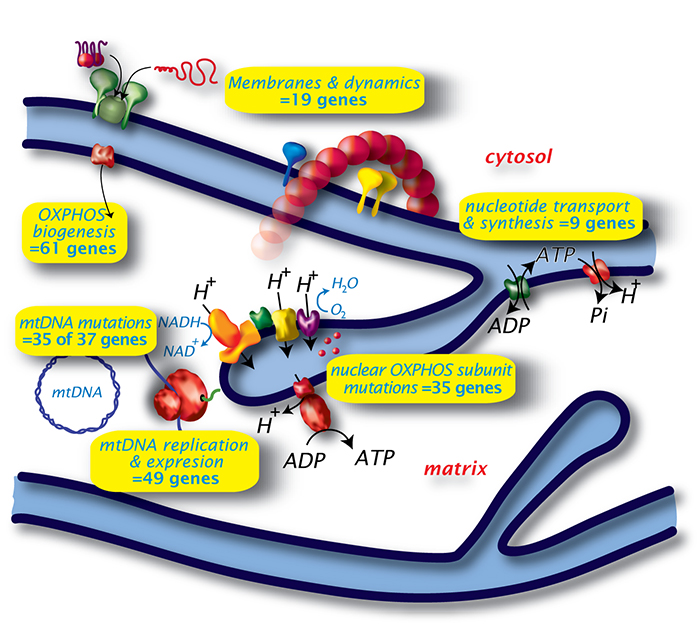
Diverse genes involved in distinct mitochondrial processes cause disease
1. Mitochondrial disease
Defects in oxidative phosphorylation (OXPHOS) result in human mitochondrial disease and affect ~1/5,000 live births. Onset can be at any age, but severe childhood disease is common and symptoms regularly involve neurological and muscular disease. Defects in respiratory chain Complex I is the most common mitochondrial disease, and results in multi-system disorders. Complex I contains 44 different protein subunits, with 7 subunits encoded by mitochondrial genes and the rest by nuclear genes. Complex I failure leads to defects in aerobic respiration with elevated lactic acid and ketone bodies. Complex I is also known to generate reactive oxygen species, an important contributor to many mitochondrial pathologies.
![]()
Mitochondrial oxidative phosphorylation and protein complexes
We are interested in understanding the function of proteins that help build the enzymes of the OXPHOS system, proteins known as assembly factors, as these are commonly mutated in mitochondrial disease. In addition, we are also interested in ways cells may be able to bypass complex I function, which may lead to the development of therapeutics to treat mitochondrial disease caused by complex I deficiency.
In collaboration with the Murdoch Children’s Research Institute at the Royal Children's Hospital, we analyse cells from patients for defects in assembly of the respiratory chain, in particular Complex I. We complement our patient studies by making knockouts in cell lines (using CRISPR/Cas9) and performing cellular, biochemical and proteomic approaches to understand the function of specific genes involved in OXPHOS.
2. Functional analysis of the human mitochondrial proteome
While we understand many aspects of mitochondrial function, we still do not fully appreciate the roles played by many individual proteins present in the organelle. Of the ~1300 proteins in human mitochondria, about 30% have no known function while another 30% have been assigned functions based on sequence similarity. We are coupling CRISPR-Cas9 gene-editing techniques to generate gene knockouts in cultured human cells and investigating how a protein's loss impacts mitochondria and the cell. This work is being coupled with state-of-the-art proteomic and bioinformatic approaches to determine how the cell changes its protein make-up to adjust to these changes. In our approach to functionalise the human mitochondrial proteome we are making new and important discoveries into fundamental aspects related to mitochondrial protein function.
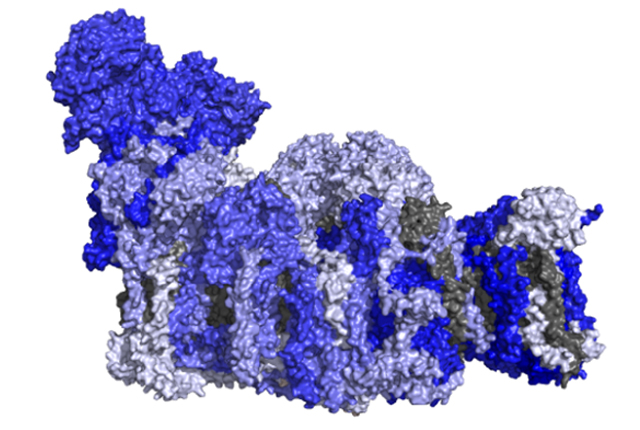
Proteomic heat-maps are correlated against known protein structures
3. Mitochondrial membrane architecture and cell death
The mitochondrial inner membrane forms highly regulated folds known as cristae that support energy production by increasing the surface area. These cristae membranes are maintained by an evolutionarily conserved complex known as the Mitochondrial Contact Site and Cristae Organizing System (MICOS). While well characterised in the yeast Saccharomyces cerevisiae, the mammalian MICOS complex is less understood. We have generated several models to analyse the MICOS complex in mammalian systems and have identified several novel proteins involved in this process.
During programmed cell death (apoptosis), mitochondrial membranes are remodelled to facilitate cytochrome c release to activate proteases that deconstruct the cell without activating the immune system. This requires remodelling of both mitochondrial membranes, suggesting that the MICOS plays an important role in this process. We are interested in how the MICOS complex contributes to this aspect of regulating cell death.

Mitochondria imaged using an electron microscope. You can see outer mitochondrial membrane and the stacks of inner membrane cristae
4. Mitochondrial dynamics
Mitochondria form a network that is finely tuned to the needs of the cell. Changes in mitochondrial fission and fusion are important events in cell signalling and development. However unscripted changes in the mitochondrial network can be pathological. Increased mitochondrial fission also appears to be a hallmark of disease states including Huntington's and Parkinson's and during apoptosis.
We have identified two new players of mitochondrial fission, MiD49 and MiD51, involved in recruiting the master fission mediator Dynamin related protein 1 (Drp1) to mitochondria. Using imaging, cell biology and structural biology, we have worked out how the MiD proteins and other members of the morphology machinery regulate mitochondrial dynamics at the cellular and molecular level. Our findings are being used to determine how fission and fusion events may be controlled in disease states. We are currently investigating the physiological consequences of MiD49/51 deficiency through animal model studies.
Mitochondria in mouse lung cells
Techniques/expertise
The laboratory has extensive knowledge in BN-PAGE, genome-editing techniques, mass-spectroscopy and advanced imaging modalities.

Protein complexes from knockout lines are often analysed by Blue-Native PAGE
Disease models
We utilise the advances in genome-editing techniques to create knockouts in vitro and in vivo, allowing us to model defects observed in patients and predict potential disease presentations where none have yet been diagnosed. This enables us to investigate the molecular underpinnings of ‘mito’, paving the way for potential future treatment.
Collaborations
We collaborate with many scientists and research organisations around the world. Click on the map to see the details for each of these collaborators (dive into specific publications and outputs by clicking on the dots).
Student research projects
The Ryan Lab offers a variety of Honours, Masters and PhD projects for students interested in joining our group. There are also a number of short term research opportunities available.
Please visit Supervisor Connect to explore the projects currently available in our Lab.
Honours and Masters Students
Honours student places are available in the Ryan lab. Our projects are designed to give students the opportunity to undertake a unique research project where they will learn a variety of techniques to skill them for the workforce and/or potential PhD positions. Experienced and friendly lab members give students daily supervision in the lab. We welcome your visit to the lab where you can meet the team.
Please contact Mike to arrange an appointment and include your academic transcript.
PhD Students
PhD positions are available to suitably qualified students with a genuine interest in our work. Applicants must have a first-class honours degree or equivalent with skills in molecular and/or cell biology.
Students should send their academic transcript and cover letter to Mike for appraisal.
PhD scholarships are awarded by Monash University on a competitive basis.