Most mRNA therapeutics require a 5' cap structure for proper cellular activity. While sequencing methods can provide indirect information about capping, they lack the specificity to precisely identify cap structures or accurately quantify capping efficiency. These are critical parameters for optimising mRNA production and ensuring therapeutic efficacy.
At the Monash RNA Mass Spectrometry Platform, we overcome these limitations through high-precision cap analysis using liquid chromatography tandem mass spectrometry (LC-MS/MS) with the RNase H method. This industry-standard approach directly characterizes cap structures and quantifies capping efficiency with superior accuracy compared to sequencing-based methods. The MS based method delivers unambiguous identification of cap variants (e.g., Cap-0, Cap-1 or Cap-2) and precise capping efficiency measurements. These are essential quality metrics for robust mRNA therapeutic development.
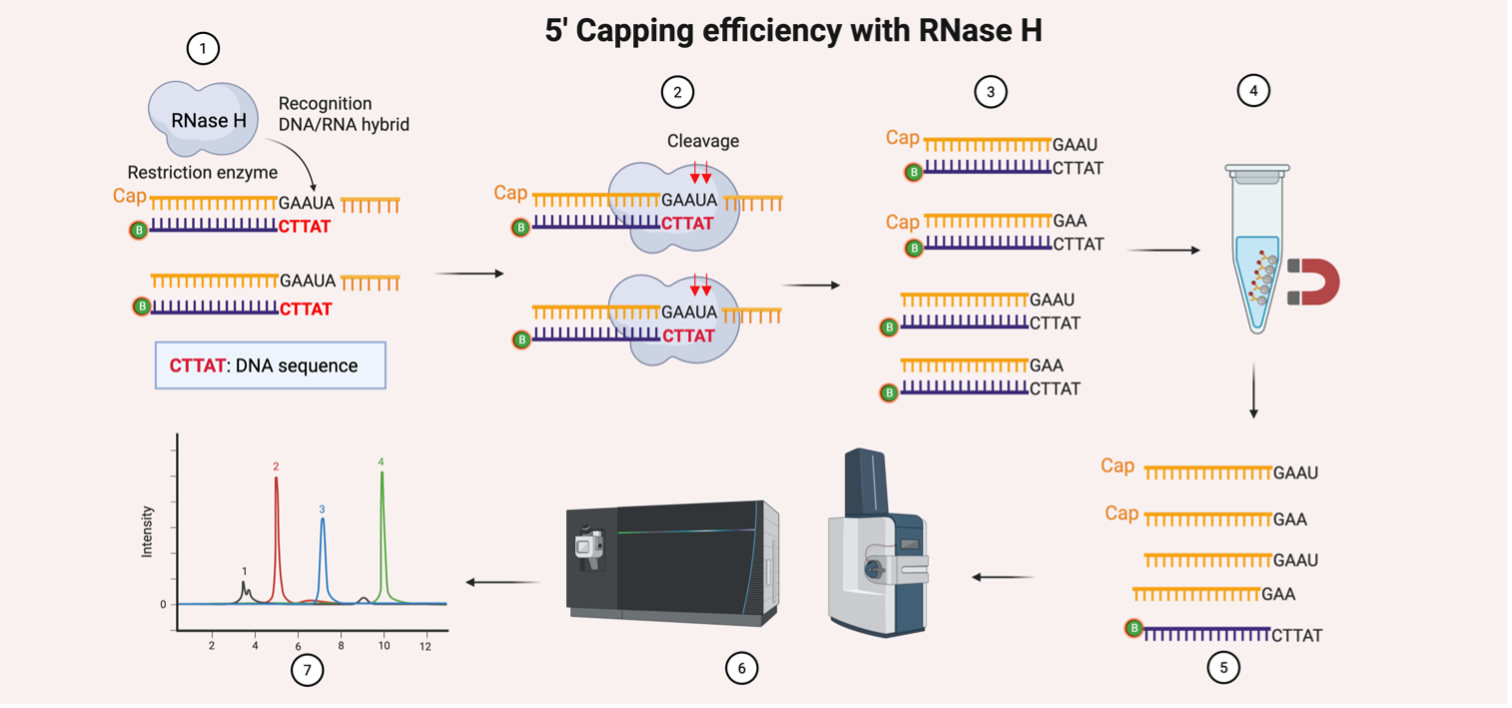 Figure 1: The quantification of 5’ capping efficiency with RNase H
Figure 1: The quantification of 5’ capping efficiency with RNase H
Heighted is the method for 5’ capping quantification [, as described by Beverly, Analytical and Bioanalytical Chemistry, 2014 ]. 1. Hybridisation: A biotinylated DNA oligonucleotide anneals to the 5’ region of the mRNA, forming a DNA:RNA hybrid; 2. RNase H Cleavage: RNase H recognises the hybrid and cleaves the RNA strand, releasing 5’ fragments of varying lengths; 3. Product Generation: Cleavage yields a mixture of capped and uncapped 5’ RNA fragments; 4. Streptavidin Bead Enrichment: The biotinylated oligonucleotide (with the bound cleaved RNA products) is captured using streptavidin magnetic beads, enriching for 5’ RNA fragments; 5. Elution: RNA fragments are eluted from the beads for downstream analysis; 6. LC-MS/MS Analysis: Eluted fragments are digested and analysed to identify and quantify cap structures (e.g., m7GpppN); 7. Data Analysis: Capping efficiency is calculated as the ratio of capped vs. total 5’ ends detected.
Polyadenylation is a critical step in mRNA manufacturing, where adenosine residues are added to the 3' end of the transcript to enhance stability and translational activity. Accurately determining poly(A) tail length distribution is essential for quality control, yet conventional methods like sequencing or biochemical assays are often costly and time-consuming. Mass spectrometry offers a superior alternative, combining speed, cost-efficiency, and high-resolution analysis. Using an optimized approach, poly(A) tails are first enzymatically released from the mRNA using RNase T1, selectively captured using oligo(dT) magnetic beads, and then analysed by a high-resolution mass spectrometer. This enables precise measurement of mass-to-charge ratios to determine the poly(A) tail length distribution, providing a robust analytical solution for mRNA therapeutic development.
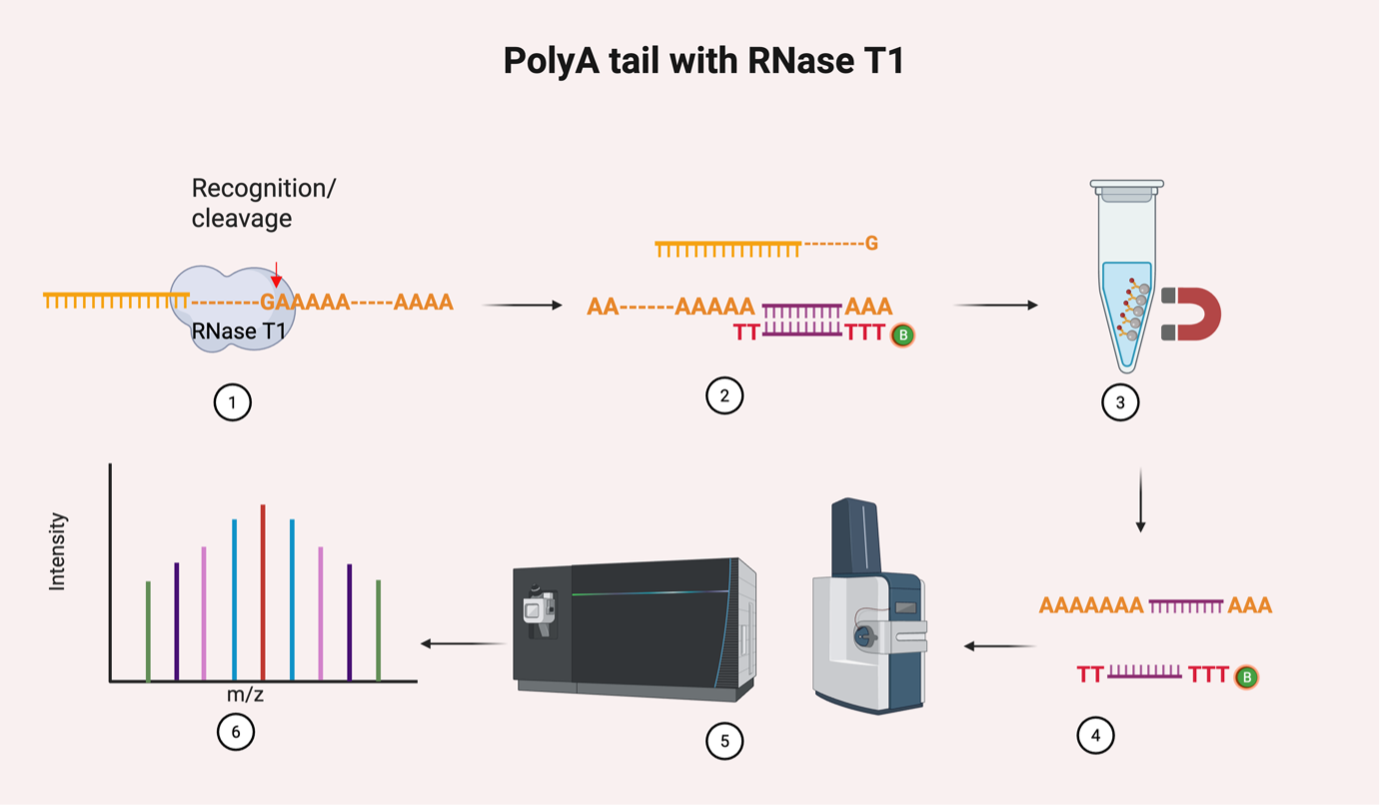 Figure 2. Quantification of poly(A) tail length determination
Figure 2. Quantification of poly(A) tail length determination
Outlined is a workflow for the quantification of poly(A) length determination. 1. RNase T1 recognition and cleavage: cleaves the RNA strand; 2. Oligo d(T) binding with the target; 3. Oligo d(T) Magnetic Beads Enrichment: The poly(A) tail is captured using oligo dT magnetic beads; 4. Elution: the Poly(A) tail is eluted from the beads for downstream analysis; 5. LC-MS/MS Analysis; 6. Data Analysis to determine the Poly(A) tail length distribution.
Over 150 chemical modifications have been identified across all RNA species—including mRNA, tRNA, lncRNA, rRNA, and microRNA. These RNA modifications playing pivotal roles in regulating gene expression in various physiological and pathological states.
Triple Quadrupole (QQQ) mass spectrometers operate most effectively in targeted mode, which allows for the highly sensitive and selective detection of modified nucleosides. This approach enables the simultaneous quantification of diverse RNA modifications in complex biological matrices. For that, enzymatic RNA digestion breaks down RNA into nucleosides, followed by LC-QQQ analysis, calibrated against synthetic standards. If absolute quantification is required, stable isotope-labelled reference standards provide internal calibration, ensuring precise measurement of the targets. The current QQQ platform provides a panel of 25 types of validated nucleoside targets, with expansion to cover additional modified nucleosides is possible based on client needs.
Recent advances in tandem mass spectrometry (MS/MS) have enabled the sequencing of full-length mRNAs, including both modified and unmodified transcripts, with near-complete coverage. RNA MS/MS sequencing can be performed rapidly, encompassing sample preparation, data acquisition, and analysis in a streamlined workflow. Beyond sequencing coverage, MS-based methods provide direct detection of RNA modifications. This represents a key advantage over next-generation sequencing (NGS), where modifications often require indirect inference.
These capabilities position RNA MS/MS as a fast and cost-effective alternative to conventional RNA sequencing. In this approach, the RNA is are partially digested into oligonucleotides, which are then applied for LC-MS/MS in negative ion mode for sequencing. The resulting spectra are matched against a reference sequence for identification.