Our research
Close
Many of the group members participate in the running of mRNA Core, a facility housed at MIPS, one of four national RNA facilities funded by Therapeutic Innovation Australia. mRNA Core collaborates with internal and external researchers who are evaluating new applications of mRNA and self-amplifying RNA (saRNA) technology. mRNA Core initially meets with collaborators to understand the project in mind and advise on a strategy for product development and testing. Subsequently mRNA constructs are designed with a view to limiting innate immune activation and maximising translational efficiency. mRNA Core can then synthesise and purify the chosen RNA molecules, and formulate as necessary for in vivo applications. Typically mRNA or saRNA molecules are encapsulated in lipid nanoparticles which are subjected to a range of quality control assays before the products are released for use by collaborators. mRNA Core prefers to operate under research collaboration agreements, implying some shared ownership of new intellectual property arising from the project. We can also provide RNA products on a fee-for-service basis as required.
mRNA Core has established projects with >25 collaborators, including projects exploring new mRNA vaccines, mRNA treatments for cancer, metabolic diseases and rare genetic diseases. Internally, mRNA Core supplies mRNA products for multiple MIPS research groups, in particular supplying products for use in the MRFF-funded CORTx program led by MIPS colleagues Angus Johnston and Natalie Trevaskis. Members of the mRNA Core team are also involved in many of the research projects described below, and supervise intern students, undergraduate and Masters placement students and PhD students.

Our experience with mRNA products began with the development of an mRNA vaccine for COVID-19. The vaccine progressed by collaboration with key researchers from The Peter Doherty Institute for Infection and Immunity into a Phase 1 clinical trial, carried out successfully in 2022 as a fourth-dose booster vaccine, using a vaccine targeted at the Beta variant of SARS-CoV-2. Our vaccine differs from the commercial ‘whole-spike’ vaccines, in that it encodes for an engineered fusion of the receptor-binding domain with the transmembrane and cytoplasmic tail of the SARS-CoV-2 spike protein. As the COVID pandemic progresses, with new variant viruses emerging on a regular basis, our RBD-TM vaccine platform has the advantage that it can overcome immune imprinting and has the potential to prevent infection of vulnerable and ageing individuals. With a view to pandemic preparedness, we are exploring the RBD-TM platform as the basis for broad-spectrum vaccines against coronaviruses. There is a very real possibility that another bat coronavirus could cross over to the human population. The SARS outbreak in 2002/2003 has been followed by sporadic outbreaks of MERS in the Middle East since 2012, both of which preceded the COVID-19 pandemic. Fortunately, neither SARS or MERS were as contagious as SARS-CoV-2, but they provide a warning that other viral infections will emerge in the future.
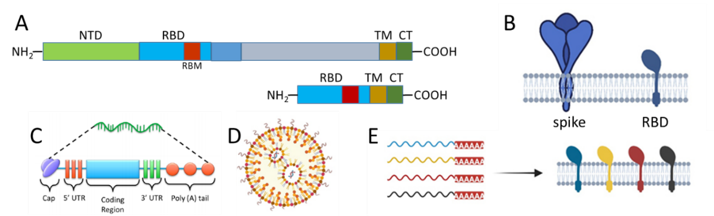
Schematic representation of combination RBD-TM mRNA coronavirus vaccines: A) whole SARS-CoV-2 spike sequence (1273 amino acids) versus RBD-TM sequence (328 amino acids); B) translated spike versus RBD; C) modified mRNA; D) lipid nanoparticle; E) RBD-TM mRNAs translated as a combination of multiple RBDs from variant coronaviruses
Lipid nanoparticle (LNP) formulations provide an elegant means to entrap nucleic acids in a particulate form. The trick is to include a weakly basic lipid, often referred to as an ionisable lipid, in the formulation. This allows the nucleic acid molecules, such as mRNA or DNA, to be drawn into the lipid particle in the presence of ethanol at low pH when the ionisable lipid is protonated and positively charged. Subsequent dialysis or diafiltration to exchange the pH and flush away ethanol results in the finished LNP product. Substantial structural changes occur within the LNP during this process. We are studying the structure of LNPs using a combination of methods including: small angle X-ray scattering, cryo electron microscopy, particle sizing and spectroscopic studies. Depending on the lipid formulation, the structure of LNPs has a profound effect on mRNA stability, mRNA entrapment, and the biological activity of the product.
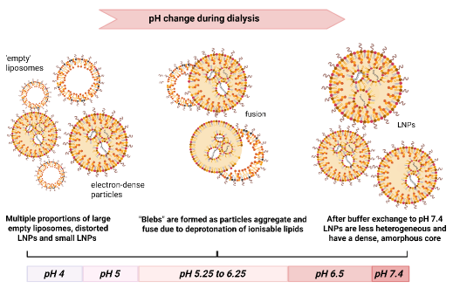
Morphological changes during manufacture of LNPs: mRNA-LNPs are generally formed at pH 4 in the presence of 25% ethanol. The removal of ethanol and buffer exchange by dialysis to produce the final product at physiological pH, results in considerable structural rearrangement. Our observations suggest that LNPs are initially polydisperse at pH 4, and contain mixed populations of particles including empty liposomes and particles containing mRNA. During buffer exchange the LNPs form partially fused (‘bleb’) structures at intermediate pH values before condensing to form a more homogeneous population of particles with dense amorphous cores.
The successful application of mRNA technology for vaccination and therapeutics depends on a thorough understanding of the biodistribution and pharmacokinetics of the formulated product, potentially to be administered by various routes (i.e. intramuscular, intravenous, subcutaneous injections, pulmonary, intranasal etc.). We study biodistribution using mRNA encoding the highly sensitive enzymatic reporter, nanoluciferase. We also collaborate with MIPS colleagues to track the fate of the lipid components within LNPs and the mRNA payload. Given that mRNA-LNP technology is relatively young, there is much to be learned before these products can be optimised for specific applications. Passive biodistribution can be influenced by changes in the formulation of LNPs, suggesting that the choice of formulation could be optimised for each therapeutic application. A major factor in the passive distribution of LNPs is their interactions with plasma components, proteins and lipoproteins, which can alter the surface properties of LNPs and mediate direct interactions with certain cell types. The classic example is adsorption of ApoE which targets LNPs to hepatocytes. The fundamental science that explains how formulation affects biodistribution is of considerable interest to several groups at MIPS. In particular we collaborate with the research groups led by Natalie Trevaskis and Angus Johnston.
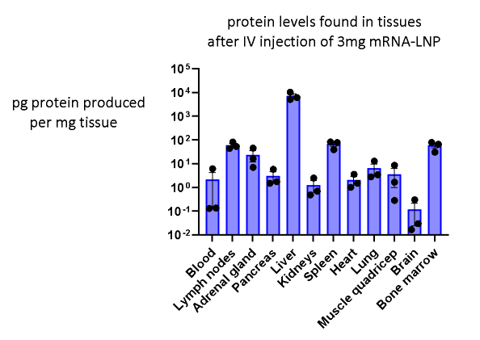
Tissue distribution of an mRNA-LNP product assayed using a reporter mRNA: Nanoluciferase can be encoded as mRNA and used to study biodistribution of formulations in vivo. In the study above, Nluc mRNA was formulated in a standard LNP then injected intravenously. The next day mice were euthanised to assay nanoluciferase translation as a function of tissue mass. The method results in a quantitative measure of protein translation in any tissue sampled.
There is growing interest in active targeting of RNA formulations to specific cell types, by incorporating targeting ligands that bind to specific extracellular proteins expressed by target cells. We collaborate with MIPS colleagues in Angus Johnston’s research group to explore antibodies and nanobodies as ligands for active targeting of LNP formulations. In this context the targeting ligand is incorporated at the surface of the LNP as a lipid conjugate. LNPs can be successfully targeted in vivo to cells of the immune system, leading to applications in treatment of cancer and autoimmune disease. We are also involved in projects targeting haematopoietic stem cells in the bone marrow using nanobodies, to allow correction of genetic blood disorders. We are exploring whether antibodies can target LNPs to the brain, and investigating the potential of lipopeptides and glycolipids as alternative targeting ligands.
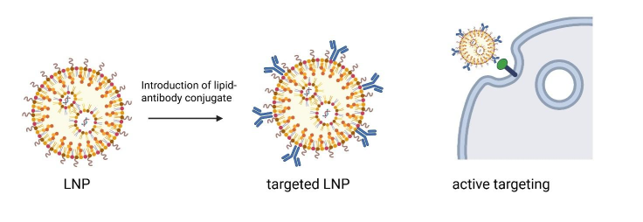
Targeted delivery of mRNA can be achieved by introducing a ligand couple to a lipid. The lipid is embedded into the surface of the LNPs resulting in display of ligands at the surface of LNPs. Using this approach, we are able to target LNPs to specific cells in vitro and in vivo.
No content
No content
Both RNA and LNPs have stimulatory effects on the innate immune system. The interplay between these effects is an important consideration for the development of safe RNA products and not surprisingly is a critical area of contemporary research. Our immune system has evolved innate responses to exposure to exogenous RNA, essentially a defence against infections by RNA viruses. These responses are regulated by a series of well-documented sensors at the surface, in the endosome, and in the cytoplasm of cells. Chemical modification of RNA can be used to reduce the innate responses. The COVID-19 vaccines made use of nucleotides containing N1-methylpseudouridine instead of uridine, to produce ‘modified mRNA’. This substitution is now widely used to reduce innate immune activation, but may not always be the most effective strategy for all indications (see above). LNPs also stimulate innate responses, perhaps due to non-specific damage rather than the specialised molecular mechanisms that sense RNA. For vaccines, some level of innate activity is essential to stimulate development of adaptive immune responses. In contrast, for therapeutic applications of RNA that are intended for chronic use, the product should be as inert as possible. We are investigating the mechanisms and extent of innate activation caused by different formulations and different routes of administration, to inform future development of RNA products for human use.
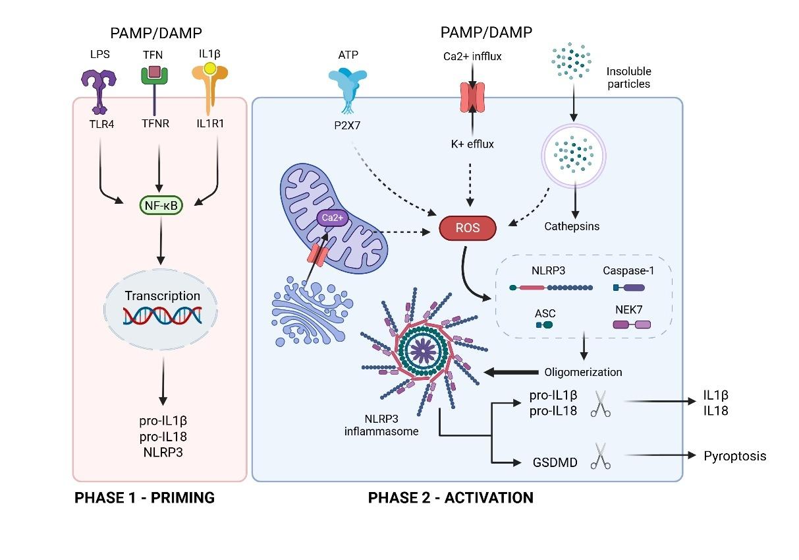
Activation of the innate immune system: The innate immune system provides a useful response to a vaccine, leading to production of an adaptive immune response. But innate immune reactions, particularly activation of inflammasomes, can be a problem for therapeutic applications of mRNA, when it would be preferable if the entire delivery system was inert. Evaluation of innate reactions is becoming an important consideration in design of mRNA delivery systems.
mRNA, and the proteins resulting from its translation, have limited half-lives in vivo. Typically peak protein production occurs over a 24 hour period after delivery of mRNA. Delivery of self-amplifying RNA can extend the duration of action but still peaks over the first few days and then declines in activity. For therapy of chronic diseases an attractive option is to use RNA to accomplish genome editing, ideally avoiding the use of viral vectors. Interest in gene editing has increased rapidly with the advances in CRISPR-Cas9-related technologies. Gene editing can take various forms, ranging from relatively simple gene knock-out, through base editing, prime editing (making use of RNA-encoded reverse transcriptase to produce DNA for integration), and potentially the objective of integrating a whole gene as cDNA into a precise location in the genome. Our group is working on the latter objective, focussing on targeted integration of cDNAs into hepatocytes in the liver, for correction of genetic diseases or provision of therapeutic proteins. Of particular interest are (i) possible mechanisms to deliver DNA to the nucleus, avoiding the need for reverse transcriptase, and (ii) characterising the incidence of off-target integration.
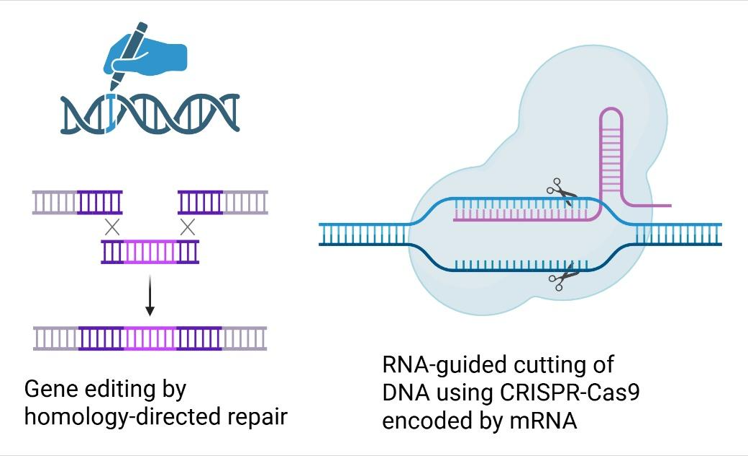
mRNA in applications of gene editing: Delivery of CRISPR-Cas9 enzymes encoded as mRNA potentially allows gene editing (i.e. gene knockout, base editing, prime editing or targeted integration of cDNA) to be achieved without requiring the use of viral vectors. We are particularly interested in introducing large pieces of cDNA to replace defective proteins, or to result in continuous expression of therapeutic proteins. This has traditionally been achieved by homology-directed repair but the advances in gene editing enzymes (e.g. CRISPR-Cas9 technology) has resulted in significant improvements in safety and efficiency, with several products in clinical development, and the approval of treatments for correction of blood disorders.