Medicinal Chemistry
We apply chemistry principles and techniques to the discovery and development of compounds to tackle disease.
.
-
Are you passionate about making a real impact in the field of pharmaceutical sciences?
In the Kaur Group, our mission is to shed light on the molecular mechanisms of diseases like dementia and pave the way for innovative diagnostic and therapeutic approaches. We are developing innovative fluorescent probes to tackle some of the most pressing challenges in healthcare today.Group website - https://www.monash.edu/mips/themes/medicinal-chemistry/research-groups/kaur
 Figure 1. Left: Fluorescent probe design for a super-resolution amyloid/GPCR sensor. Right: Fluorescent probes can be used to (a) identify amyloid biomarkers (b) visualise biomolecules and structures at the nanoscale, shown is a comparison of traditional vs super-resolution image of microtubules and (c) report on the hydrophobicity of the surfaces of biomolecules that determine their molecular interactions.
Figure 1. Left: Fluorescent probe design for a super-resolution amyloid/GPCR sensor. Right: Fluorescent probes can be used to (a) identify amyloid biomarkers (b) visualise biomolecules and structures at the nanoscale, shown is a comparison of traditional vs super-resolution image of microtubules and (c) report on the hydrophobicity of the surfaces of biomolecules that determine their molecular interactions.1. Unravelling the Mysteries of Dementia
Imagine being at the forefront of developing a diagnostic tool that could revolutionise how we detect and understand dementia. As part of our team, you'll work on the synthesis and characterisation of fluorescent probes that can identify specific biomarkers and stage of
Alzheimer’s Disease (AD). Our first-generation AD diagnostic (Figure on the right) discriminates 6- and 12-month-old wildtype (WT) and AD (APP/PS1) mice. This ground-breaking research has the potential to transform early diagnosis and treatment strategies for millions of people worldwide. See our research featured on 7News.
2. Illuminating the Nanoscale World
Dive into the fascinating realm of super-resolution imaging! Super-resolution microscopy has transformed our ability to study biomolecules at the molecular level. This Nobel Prize-winning technology offers immense potential for understanding molecular interactions. However, its full capabilities are constrained by the scarcity and suboptimal photophysical properties of available fluorophores, limiting the quantitative information researchers can obtain. Our projects focus on designing fluorescent probes that can reveal the intricate nanoscale structures of amyloids and GPCRs (G protein-coupled receptors). This research opens up new possibilities for understanding disease mechanisms at a molecular level and could lead to breakthroughs in drug discovery and personalised medicine.
3. Sensing the Unseen: Biological Microenvironments
While fluorescent probes are typically developed to report on structures, there are hardly any fluorescent probes that explore the hidden world of cellular microenvironments. Our research involves developing fluorescent probes that can detect local changes in polarity and viscosity within biological systems. This exciting work bridges the gap between chemistry and biology, offering insights into how these subtle environmental changes relate to progression of Alzheimer’s Disease.
What you will gain
In addition to being a part of a dynamic team pushing the boundaries of what's possible in fluorescent probe technology and its applications in healthcare, you will :
- gain experience in synthetic organic chemistry
- work with state-of-the-art imaging techniques and probe design methodologies
- undertake an interdisciplinary approach combining chemistry, biology, and microscopy
Key publications
1. N. Trinh, K. Bhuskute, N.R. Varghese, J.A. Buchanan, Y. Xu, F. M. McCutcheon,…A. Kaur, A Coumarin-Based Array for the Discrimination of Amyloids. ACS Sensors 2024, 9, 2, 615–621.
2. A. Kaur, L. D. Adair, S. R. Ball, E. J. New, M. Sunde, A Fluorescent Sensor for Quantitative Super-Resolution Imaging of Amyloid Fibril Assembly. Angewandte Chemie Int. Ed. 2022, 61, e202112832.
3. K. Kikuchi, L. D. Adair, J. Lin, E. J. New, A. Kaur, Photochemical Mechanisms of Fluorophores Employed in Single-Molecule Localization Microscopy. Angewandte Chemie. Int. Ed. 2023, 62, e202204745.
Contact
Leave this here so that Accordion nested does not detect this CT as not existing. -
Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences
The Capuano group’s research interests focus on the chemical synthesis and pharmacological evaluation of allosteric modulators targeting G protein-coupled receptors (GPCRs) as new treatment strategies for neurological and neurodegenerative disorders.
The group is currently working on the development of both negative and positive allosteric modulators of the class A GPCRs, namely the dopamine D2 receptor (pictured left [1]) and the M4 muscarinic acetylcholine (ACh) receptor, as novel mechanisms of action for the treatment of schizophrenia, a debilitating mental disorder that affects ~1% of the world’s population. Current therapeutics act as dopamine D2 receptor orthosteric antagonists that provide only partial symptom relief and often trigger prominent side effects. Targeting the allosteric site provides an alternative safer treatment option with potentially better therapeutic outcomes and reduced side-effect liability.

In addition, the class C GPCR, the metabotropic glutamate 5 (mGlu5) receptor (pictured centre [2]), has a vast disease prevalence due to its significant involvement in excitatory neurotransmission and synaptic plasticity. As such, the mGlu5 receptor has become a valuable therapeutic target over recent years for neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson's disease (PD), Huntington’s disease (HD) and Amyotrophic Lateral Sclerosis (ALS/MND).
Other research areas include applying chemical biology approaches towards the development of molecular probes (e.g. fluorescently-labelled molecules) for the elucidation of relevant molecular pharmacology of ‘hit’ and ‘lead’ compounds of interest, and visualising cellular events (pictured right: dopamine D2R cells labelled with an in-house probe)
The research projects are multi-disciplinary in nature and will expose you to state-of-the-art laboratories for chemical synthesis, computational chemistry techniques and expertise relevant to drug design, modern chromatography & spectroscopy facilities for compound purification and characterisation, in addition to contemporary analytical pharmacology laboratories for biochemical evaluation of novel drug candidates.
References
[1] D. Im et al. Structure of the dopamine D2 receptor in complex with the antipsychotic drug spiperone. Nature Comm. 11, 6442 (2020).
[2] C. Nasrallah et al. Agonists and allosteric modulators promote signaling from different metabotropic glutamate receptor 5 conformations. Cell Reports 36, 109648 (2021).
Contact
Leave this here so that Accordion nested does not detect this CT as not existing. -
Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences
The Flynn group works in three key areas: the development of new synthetic methods to facilitate drug discovery, chemical biology and drug design (Figure 1)

Organic synthesis
Nature has provided significant inspiration in the design of potent and selective pharmacophores. Bioactive natural products that target proteins very often contain many sp3-carbons and associated stereocentres, affording them 3D structures that bind potently and selectively to the 3D binding domains of proteins. On the other hand, natural products that bind to polynucleotides (DNA or RNA) very often have many sp2-carbons and heteroatoms, forming π-rich surfaces that interact with nucleotide base pairs (eg intercalators). Our research in organic synthesis is generating new methods for the rapid assembly of sp3- and sp2-rich polycyclic scaffolds for targeting proteins and polynucleotides, respectively. A key feature of these chemistries is the use of diversity-oriented and multi-bond forming reactions to simplify the synthesis and facilitate diversification of the scaffolds.
Chemical Biology and Medicinal Chemistry
Employing our novel chemistry and other synthetic methods we have generated biased libraries for target-based and phenotypic-based drug discovery. In our subsequent drug design we also incorporate design elements that help direct delivery of the drug to specific tissues/organs.
Target-based drug discovery and directed drug distribution
GPCR targets: sphingosine-1-phosphate-receptor-1 (S1P1) degraders that accumulate in mesenteric lymph-nodes, as a safe and effective treatment of inflammatory bowel disease. Protease-activated receptor 2 negative allosteric modulators (PAR2-NAMs) for the treatment of pulmonary and liver fibrosis and associated organ failure
Kinase targets: Sphingosine-kinase-1 (SK1) degraders for the treatment of pulmonary arterial hypertension and cancer (spinout company formed: Ankere Therapeutics). Tissues restricted kinase (eg ALK5 and ROCK2) inhibitors with antifibrotic effects that concentrate in the target tissue (lung, gut or liver).
Epigenetic modulators: In this program we are generating molecules that degrade key proteins and polynucleotide targets involved in aberrant gene transcription for the treatment of various cancers.
Phenotypic-based drug discovery for unmet medical needs
Antifibrotic and antisteatotic drugs: by developing screens of fibroblast activation and lipid-induced steatosis (accumulation of fats in hepatocytes) we have discovered new drug leads, including potent antifibrotic therapy (clinical candidate) that has formed the basis of a spinout company Cincera Therapeutics.
Contact
Leave this here so that Accordion nested does not detect this CT as not existing. -
Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences
Innovations in synthetic chemistry have had a profound impact over the past century enabling the discovery and development of life-changing medicines and medical imaging agents. Click chemistry has emerged as a key strategy in synthetic chemistry to overcome the limitations of traditional chemical synthesis (e.g. time consuming, labour intensive) using high energy spring-loaded reactants to forge complex new molecules.
While click chemistry was initially conceived to streamline the drug discovery process, it is now used in almost every corner of chemistry, materials science, and biology, and the significant impact of this approach was recognised in 2022 with the Nobel prize in Chemistry. However, the potential of click chemistry to build diverse, complex molecules with ever increasing economy, efficiency, and precision, remains significantly underexplored.
The Smedley group aims to reinvigorate the wider family of click chemistry reactions using a novel Diversity Oriented Clicking (DOC) approach; a powerful strategy for the modular synthesis of complex molecular frameworks. DOC uses spring-loaded connector molecules (DOC connectors) capable of undergoing a variety of thermodynamically driven click chemistry transformations to access valuable molecules that are difficult or impossible to prepare using existing technologies.
Projects in this area will explore the use of novel DOC connector molecules to access diverse heterocyclic frameworks. These frameworks are highly sought after in medicinal chemistry and can also serve as tool compounds for deciphering biological mechanisms to aid in drug discovery and design.
Through undertaking research in this group, candidates will have the opportunity to work in and develop skills in the following areas:
- Synthetic organic/medicinal chemistry;
- Photochemical probe development;
- Design covalent warheads for biological target identification;
- Late-stage drug functionalisation; and
- Analytical techniques (NMR, mass spectrometry, HPLC, LCMS).
Key Publications
- Smedley, C. J., Li, G., Barrow, A. S., Gialelis, T. L., Giel, M.-C., Ottonello, A., Cheng, Y., Kitamura, S., Wolan, D. W., Sharpless, K. B. and Moses, J. E., Diversity Oriented Clicking (DOC): Divergent Synthesis of SuFExable Pharmacophores from 2-Substituted-Alkynyl-1-Sulfonyl Fluoride (SASF) Hubs. Angew. Chem. Int. Ed., 20202020, 59, 12460-12469.
- Smedley, C. J.,A diversity oriented clicking strategy: the stereoselective synthesis of highly-functionalised olefins from 2-substituted-alkynyl-1-sulfonyl fluorides.Chem. Commun, 2022, 58, 11316-11319.
- Cheng, Y., Li, G., Smedley, C. J., Giel, M.-C., Kitamura, S., Woehl, J. L., Bianco, G., Homer, J. A., Cappiello, J. R., Wolan, D. W., Moses, J. E. and Sharpless, K. B., Diversity oriented clicking delivers β-substituted alkenylsulfonyl fluorides as covalent human neutrophilelastase inhibitors. Proc. Natl. Acad. Sci. U.S.A.,2022, 119, e2208540119.
- Barrow, A. S., Smedley, C. J., Zheng, Q., Li, S., Dong, J. and Moses, J. E., The growing applications of SuFEx click chemistry. Chem. Soc. Rev., 2019, 48, 4731-4758.
- Zheng, Q., Woehl, J. L., Kitamura, S., Santos-Martins, D., Smedley, C. J., Li, G., Forli, S., Moses, J. E., Wolan, D. W. and Sharpless, K. B., SuFEx-enabled, agnostic discovery of covalent inhibitors of human neutrophil elastase Proc. Natl. Acad. Sci. U.S.A., 2019, 116, 18808-18114.
Contact
Leave this here so that Accordion nested does not detect this CT as not existing. -
Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences
Projects involve the use of a range of emerging technologies to drive the discovery of new synthetic strategies. These strategies will be used to access novel molecular frameworks that can then be utilized in the development of new therapeutic candidates. Therapeutic focus areas include cancer, infectious disease and cardioprotection.

Contact
Leave this here so that Accordion nested does not detect this CT as not existing. -
Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences
Our group focuses on developing peptides as human therapeutic leads for a range of diseases, as well as bioinsecticides.
1. We have a strong focus on peptides that target the voltage-gated potassium channel Kv1.3, as these are highly promising therapeutic leads for the treatment of autoimmune diseases such as psoriasis, rheumatoid arthritis and inflammatory bowel disease, and neuroinflammatory diseases such as Alzheimer’s and Parkinson’s diseases.1, 2

In order to advance these peptide leads to the clinic we are pursuing a number of strategies:
- Lipidation to enhance circulating lifetime and modulate biodistribution
- Modifications to enhance uptake into the central nervous system
- Modifications to improve therapeutic efficacy and generate oral bioavailability
- PET and fluorescence imaging to monitor biodistribution
- Evaluation in new disease models
2. The potent and selective peptide inhibitors of Kv1.3 that we are developing have come from sea anemones and scorpions. With collaborators in Queensland, we continue to identify, make and test novel peptides from Australian sea anemones with the aim of finding new therapeutic candidates or bioinsecticides.3, 4
3. Novel inhibitors of Type 2 diabetes (T2D). We are developing inhibitors of an enzyme known to play key roles in β-cell failure and insulin resistance. We have identified a number of peptides that inhibit the interaction between PKCε and RACK2.5, 6 This project will focus on enhancing the potency and cell membrane permeability of these peptides as drug leads and subsequently testing them in animal models of T2D.
PhD and Honours projects are available in all of these areas.
Our research projects use the following techniques:
- Peptide chemistry
- Structural biology
- NMR spectroscopy, mass spectrometry, HPLC
- Molecular modelling, molecular dynamics, docking
- Peptide and protein expression, purification and characterisation
- Cell permeability and cytotoxicity assays
References
- Tajti, G., et al. (2020) Biochem. Pharmacol. 181, 114146.
- Wai, D. C. C., et al. (2022) Bioconjug Chem 33, 2197-2212.
- Prentis, P. J., et al. (2018) Toxins 10, 36.
- Elnahriry, K. A., et al. (2024) Biochim Biophys Acta Proteins Proteom 1872, 140952.
- Schmitz-Peiffer, C. (2020) Trends Endocrinol. Metab. 31, 344-356.
- Chandrashekaran, I. R., et al. (2018) FEBS Lett. 592, 179-189.
Contact
Leave this here so that Accordion nested does not detect this CT as not existing. -
Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences
The Chalmers research group uses molecular modelling and synthetic medicinal chemistry techniques to develop new peptide-nanomaterials, biologically active peptides and small molecules. We use molecular modelling to investigate peptide structure, how drugs or peptides interact with proteins (for example, how peptides bind to receptors) and how molecules interact with cell membranes. We also use molecular simulations to model the behaviour of drug formulations within the body. Information from our computational models is used to evaluate drug formulations and to design and synthesise new bioactive compounds for testing.
 Molecular dynamics simulation of a peptide binding to the neurotensin-1 receptor.
Molecular dynamics simulation of a peptide binding to the neurotensin-1 receptor.Research projects are available in the following areas:
- Peptide nanomaterials
- Bioactive peptides
- Computational drug formulation
- Machine-learning and artificial intelligence
Our research projects use the following techniques:
- Molecular modelling: Molecular dynamics, docking, free energy calculations
- Medicinal chemistry: Solid phase peptide synthesis. Small molecule synthesis. Developing structure-activity relationships
- Analytical methods: NMR spectroscopy, HPLC/MS, membrane permeation assays.
- Machine learning: Neural network models for molecular and peptide design
Key publications
Williams-Noonan … Chalmers, Yarovsky, Membrane Permeating Macrocycles: Design Guidelines from Machine Learning. Journal of Chemical Information and Modeling 2022, 62 (19), 4605-4619.
Guruge, Warren, Benameur, Pouton, Chalmers. Aqueous phase behavior of the PEO-containing non-ionic surfactant C12E6: A molecular dynamics simulation study. Journal of Colloid and Interface Science 2021, 588, 257-268.
Contact
Leave this here so that Accordion nested does not detect this CT as not existing. -
Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences
Our research uses synthetic organic chemistry to combine and functionalise biomolecules (peptides, proteins, nucleic acids) and their mimics in inventive ways to create controllable therapeutic agents. We have a particular focus on developing and applying cutting-edge synthetic protein methodologies to assemble and decorate these complex biomolecules. We also use designer mimics of nucleic acids in combination with proteins to create smart, responsive systems which can release or activate therapeutic agents specifically at disease sites for improved safety and toxicity profiles.
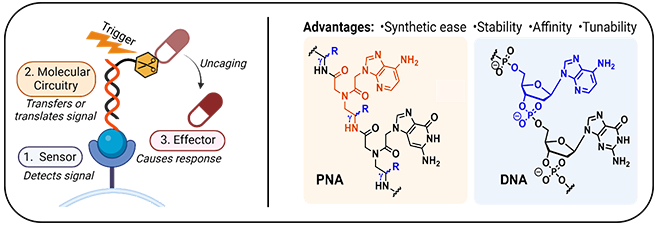
Contact
Leave this here so that Accordion nested does not detect this CT as not existing. -
Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences
In in-silico drug discovery, the goal is to find promising molecules (hits) quickly and efficiently. This project focuses on building virtual screening engines powered by quantum descriptors and AI predictors to evaluate thousands (or millions) of drug-like molecules.
You’ll also work with uncertainty-aware AI models (e.g., Bayesian neural networks) to prioritize the most promising molecules, and help develop tools to filter and rank molecules based on both chemical intuition and rigorous computation.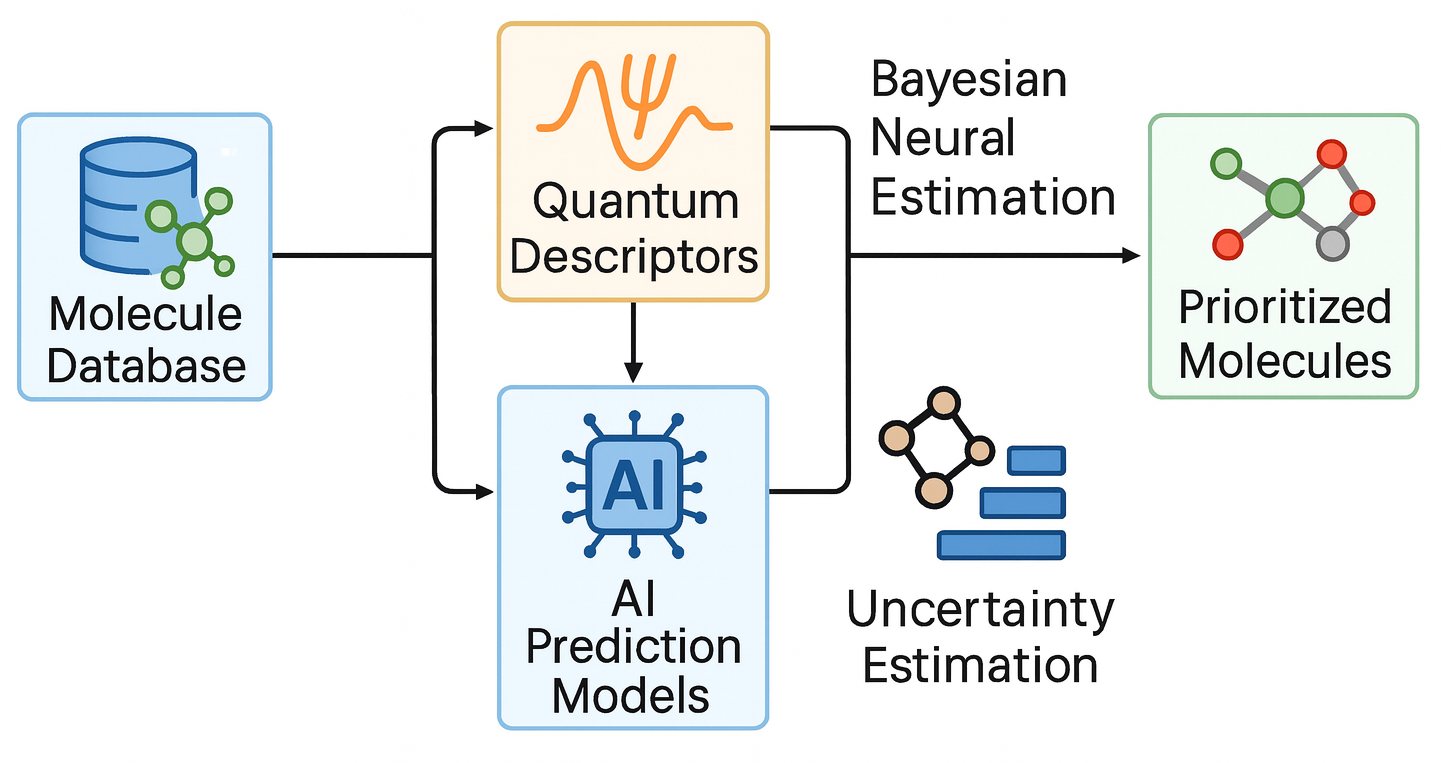
Skills You’ll Learn
- High-throughput molecular screening workflows
- Quantum-derived scoring functions
- Bayesian machine learning and ensemble models
- Tools for drug-likeness, ADMET prediction
Applications
- Hit identification for undruggable targets
- Lead filtering with quantum-accurate models
- Reducing false positives in virtual screening
References
- Deep learning pipeline for accelerating virtual screening in drug discovery. https://doi.org/10.1038/s41598-024-79799-w
- Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery. https://doi.org/10.1021/acscentsci.0c00229
- Boltz-2: Towards Accurate and Efficient Binding Affinity Prediction. https://doi.org/10.1101/2025.06.14.659707
Contact
Leave this here so that Accordion nested does not detect this CT as not existing. -
Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences
Imagine designing molecules from scratch that are guaranteed to be drug-like and tailored to bind a target. In this project, you’ll develop generative AI models (e.g., diffusion models, autoencoders, transformers) to optimize molecular structures starting from an initial lead compound.
You’ll guide the generative AI using protein structure, binding affinity, reactivity, and other constraints—either predicted by ML or computed from quantum chemistry. This project is ideal if you enjoy combining creativity, chemistry, and AI to solve hard problems in drug design.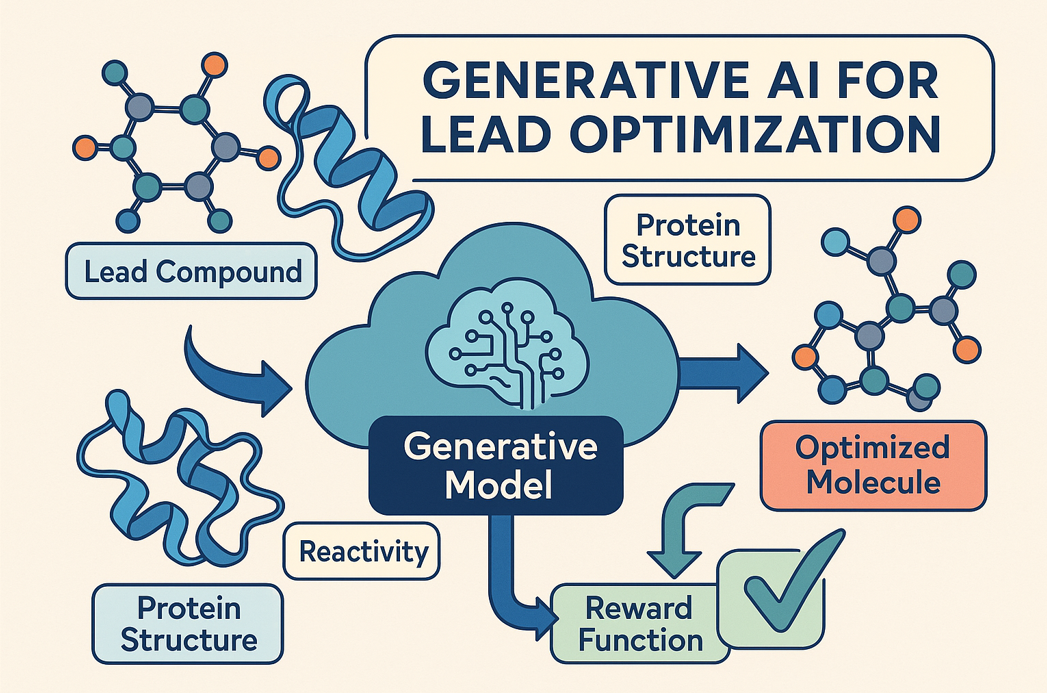
Skills You’ll Learn
- Building generative models for molecules
- Lead optimization via AI-guided exploration
- Using reward functions (e.g., for binding, toxicity)
- Learning from quantum and medicinal chemistry data
Applications
- Improving lead compounds for higher binding affinity
- Designing drugs with better ADMET properties
- Automating multi-objective drug optimization
References
- Equivariant Diffusion for Molecule Generation in 3D. https://arxiv.org/abs/2203.17003
- Deep Lead Optimization: Leveraging Generative AI for Structural Modification. https://doi.org/10.1021/jacs.4c11686
- GuacaMol: Benchmarking Models for de Novo Molecular Design. https://doi.org/10.1021/acs.jcim.8b00839
Contact
Leave this here so that Accordion nested does not detect this CT as not existing. -
Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences
What if we could predict how a molecule behaves and how confident we are in that prediction—before ever making it in the lab?
In this project, you’ll help build the next generation of QSAR (Quantitative Structure–Activity Relationship) models—ones that don’t just predict whether a molecule is active, soluble, or toxic, but also estimate how likely those predictions are to be correct. This means you won’t just say “this looks promising”, but “this looks promising—and we’re 90% confident about it.”To do this, you’ll train AI models that combine:
- Experimental data (like pIC₅₀, LogP, or toxicity flags),
- Quantum mechanical descriptors (e.g. frontier orbital energies, dipoles, ESPs),
- And bioisosteric transformations (e.g. methyl → fluoro, phenyl → pyridyl) that chemists use every day to tune molecules.
You’ll use your models to explore how specific structural changes affect molecular properties across multiple endpoints. You’ll also estimate the uncertainty in those changes—so you can prioritize substitutions that are most likely to succeed, and know when you're entering new chemical territory.
Rather than making hundreds of molecules and hoping a few work, your models will help focus experimental testing on the most promising (or most informative) candidates—saving time, money, and lab effort.
This project is perfect if you're interested in AI, chemistry, and real-world drug discovery—and want to work at the intersection of all three.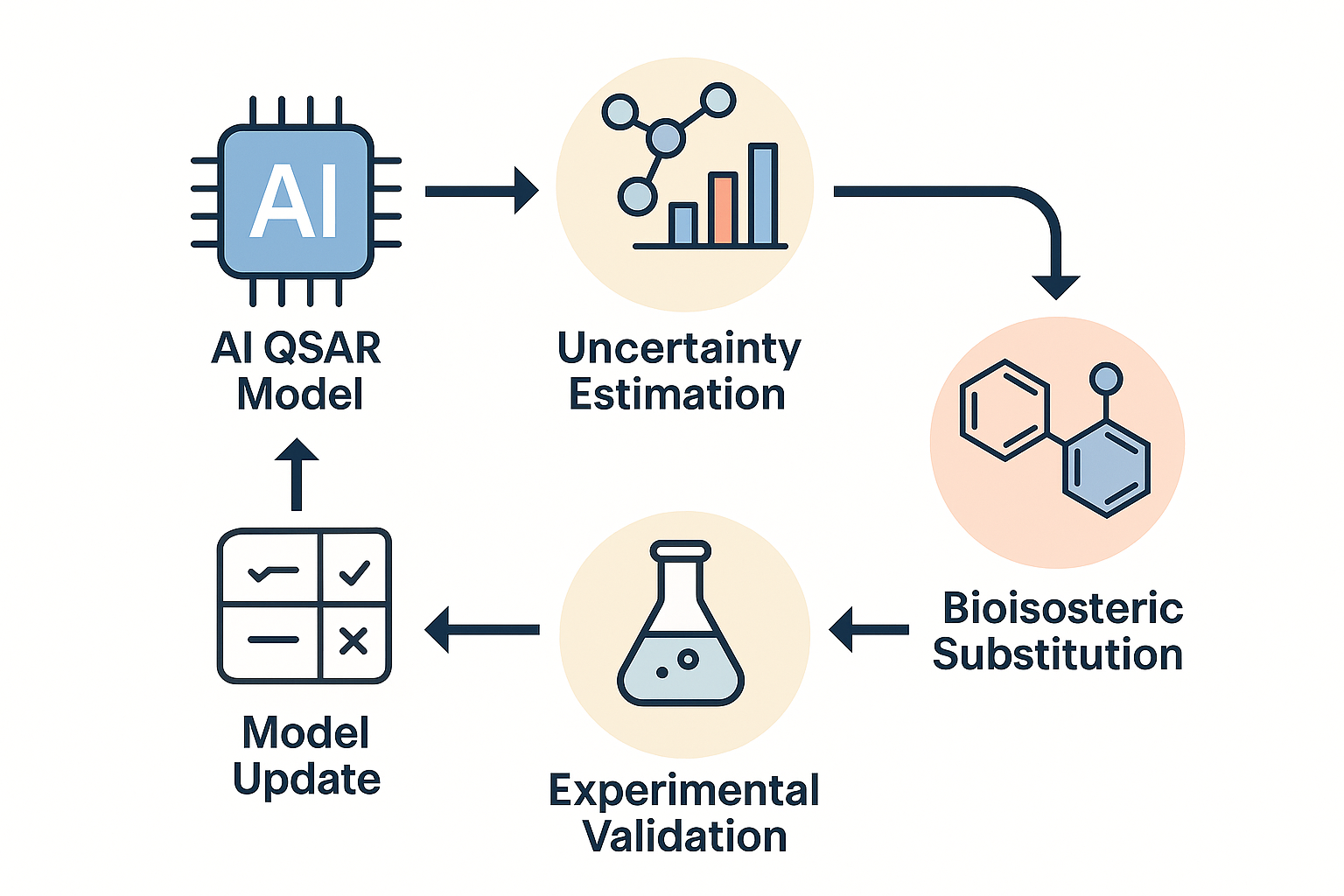
Skills You’ll Learn
- How to build modern QSAR models with machine learning (regression + classification)
- Generating quantum chemical descriptors with tools like xTB or Psi4
- Quantifying uncertainty using deep ensembles or Bayesian methods
- Matched Molecular Pair (MMP) analysis and how to represent chemical transformations
- Multi-objective optimization across properties like activity, lipophilicity, and toxicity
- Using cheminformatics tools like RDKit to handle real molecular data
What You’ll Apply This To
- Predicting the effect of small structural changes on molecular properties
- Evaluating and suggesting new bioisosteric substitutions
- Helping prioritize which compounds should be synthesized and tested
- Understanding when your model is confident—and when it's just guessing
References
- Bioisosterism: A Rational Approach in Drug Design. https://doi.org/10.1021/cr950066q
- Comprehensive strategies of machine-learning-based quantitative structure-activity relationship model. https://www.cell.com/iscience/fulltext/S2589-0042(21)01020-8
- Characterizing Uncertainty in Machine Learning for Chemistry. https://doi.org/10.1021/acs.jcim.3c00373
Contact
Leave this here so that Accordion nested does not detect this CT as not existing. -
Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences
The Jörg lab focuses on the development of novel chemicals probes for a range of different disease areas. The molecules we design support studies to improve our understanding of proteins on a molecular level, and to address important questions in molecular biology related to the structure, activation, degradation, oligomerisation, allostery and selectivity of selected drug targets. By developing first-in-class chemical probes of therapeutically relevant proteins, we will get pivotal insights into the action of drugs, drug targets and diseases that will enable the development of improved therapeutics and diagnostics. In addition, our lab is also interested in the area of sustainable drug discovery and DNA-encoded libraries.
Consider joining the Jörg lab if you are interested in the following research areas:

Design, Synthesis and Evaluation of Novel Multifunctional Fluorophores
Fluorescently-labelled small molecules are powerful tools which utilise the concept of photoluminescence to visualize and investigate drug targets. Fluorescent probes offer a safe alternative to radioligands and demonstrate enormous versatility for ligand binding, high-throughput screening and real-time measurement of ligand-induced receptor trafficking. This has driven significant innovation in the development of sensitive fluorescent probes and platform technologies. Further, small molecule-based fluorescent probes often provide superior properties over endogenous reporters such as fluorescent proteins due to their brightness, photostability, Stokes shift and high quantum yields.Despite the advance in fluorescence technologies, there are still limitations to these approaches due to the lack of fluorophores with desired chemical and photophysical properties. Our project addresses this critical knowledge gap by developing novel multifunctional fluorophores, which can be applied to study ‘click-to-release’ cancer therapies, dimerization of GPCRs and protein degradation. Therefore, this focuses on the design, synthesis and evaluation of first-in-class multifunctional reduction-sensitive and/or turn-on fluorophores.
Design, Synthesis and Evaluation of Protein Degraders and Fluorescent Probes
Protein degraders and fluorescent probes are chemical modalities that have a wide range of potential applications in drug discovery and chemical biology. The development of chemical probes is crucial to obtain insights into the mode-of-action of drugs, as well as the role of proteins in health and diseases. Hence, our group is interested in the design, synthesis and evaluation of novel selective chemical probes to 1) protein degradation, (2) receptor function and dimerization, and (3) drug delivery systems.
More specifically, we have projects in the following areas:
- Development of protein degraders, such as PROTACs, CLIPTACs and degron-TAGs to assist studies of protein function and target deconvolution
- Development novel fluorescent probes for a range of G protein-coupled receptors to study receptor function and receptor dimerization
- Development of chemical probes to study antibody delivery systems
Drug design beyond the patient: Mapping non-human pharmacology to probe the environmental risk of opioids and/or beta-blockers
Collaborative Project with Aili Langford & Lauren May
Pharmaceuticals and lifestyle drugs are increasingly considered contaminants of concern for environmental and ecological health. Active pharmaceutical ingredients and metabolites are increasingly found in soils, biota, sediments, surface water, groundwater and drinking water. G protein-coupled receptors (GPCRs)-targeted pharmaceuticals, make up around 34% of all FDA approved pharmaceuticals target. Amongst other GPCR drugs, opioids, beta-blockers and their metabolites are frequently detected in wastewater streams globally. These drugs target the GPCR orthosteric binding site recognised by the endogenous agonist, similar to the vast majority of FDA-approved GPCR-targeted pharmaceuticals. Orthosteric binding pockets shared by a common endogenous agonist are typically conserved across species. However, the binding affinity of human pharmaceuticals has not been systematically mapped across non-human species. Our project addresses this critical knowledge gap, mapping concentrations of contaminant pharmaceuticals in the environment with species most likely to be at risk.
Therefore, this project will identify at-risk non-human species by mapping the evolution of drug binding pockets for select FDA-approved beta-blockers, opioids and their metabolites that persist in the environment. Validation of computational approaches to predict non-human pharmacology at the different adrenergic and opioid receptor subtypes will enable ethical and rational selection of indicator species for detailed ecotoxicological profiling. As opioids and beta-blockers use continues along its upward trajectory, environmental risk minimisation of these drugs will be pivotal. Long-term, these models might be applied to structure-based design of novel drugs interacting with receptor regions lacking evolutionarily conservation.
Diversity, equity and inclusion and cultural awareness as part of curricula
Teaching focused project in collaboration with Betty Exintaris & Nilushi Karunaratne
While diversity, equity and inclusion (DEI)as well as cultural awarenessare core values of most tertiary education institutions, this is often not reflected in the teaching curriculum. Therefore, in this project we will explore the international landscape for DEI initiatives within the tertiary education sector. Furthermore, we will analyse the course curricula at MIPS with the aim to highlight opportunities to implement DEI and cultural awareness aspects related to pharmacy and drug discovery. Lastly, this project will facilitate the development of novel methods to teach important DEI and cultural aspects of pharmacy and drug discovery, which are inadequately addressed with the current curriculum at MIPS and more generally in pharmacy and pharmaceutical science education. Ultimately, this research will ensure that future pharmacists and drug discovery scientists will have a broader perspective and increased awareness of the different medical needs and/or access to medical services as well as drugs that may affect people differently.
Contact
Leave this here so that Accordion nested does not detect this CT as not existing. -
Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences
Research in my group operates at the early stages of drug discovery projects. We first identify small molecules that bind to a therapeutic target of interest. This provides “hit” compounds, which we subsequently elaborate to improve their binding affinity to generate more potent “lead” candidates.
The hits are identified by using either fragment-based screening or DNA-encoded library screening. Hits are validated and their binding to the target is verified using a suite of different biophysical binding assays. We use X-ray crystallography and NMR spectroscopy to generate structures of hit compounds in complex with their targets. We then use this information to design and synthesise new compounds with improved affinity.
We have developed a method to accelerate these cycles of “Design-Make-Test”. This involves parallel microscale synthesis and testing of libraries of compounds. We use computational chemistry to optimise the chemical diversity of the libraries, robotics to perform the synthesis, and we have developed methods to screen the resulting compounds without the need for extensive purification. This means that we can design and test hundreds of compounds and accelerate the hit-to-lead process.
 Figure 1: Structural and biophysical characterisation of protein-ligand binding. (A) The binding pose of a hit compound identified by X-ray crystallography. (B) Concentration-dependent binding measured by surface Plasmon resonance (SPR). (C) Analysis of SPR data to determine affinity.
Figure 1: Structural and biophysical characterisation of protein-ligand binding. (A) The binding pose of a hit compound identified by X-ray crystallography. (B) Concentration-dependent binding measured by surface Plasmon resonance (SPR). (C) Analysis of SPR data to determine affinity.We have several projects where this approach is being applied across a range of different therapeutic targets
- Anti-infective agents targeting Gram-negative bacteria. We have developed compounds that inhibit protein folding in the periplasm of Gram-negative bacteria. They do so by inhibiting an enzyme called DsbA that catalyses the formation of disulfide bonds in newly synthesised proteins. Our DsbA inhibitors work in two distinct ways. Firstly, they inhibit virulence by preventing bacteria from producing the proteins that it uses to cause disease. Secondly, they inhibit some of the mechanisms that bacteria use to gain resistance to current antibiotics. In this way they can re-sensitise drug resistant bacteria to antibiotics.
- Inhibitors of flaviviral enzymes. Flaviviruses such as Dengue, Japanese encephalitis and Zika infect hundreds of millions of people each year. Dengue alone is estimated to cause almost 400 million infections each year. Although individual flaviviruses differ in their pathogenesis, their underlying replication machinery is conserved. Enzymes that are encoded in the flaviviral genome, including the protease and the RNA-dependent RNA-polymerase are highly conserved and essential for viral replication. Therefore, we are targeting these enzymes to develop broad-spectrum inhibitors as drug candidates for current and emerging flavivirus-borne diseases.
- Modulators of lipid signalling. Lipid signalling plays a key role in the pathology of many diseases, including diabetes, atherosclerosis and cancer. We are developing small molecule modulators of several proteins that have a role in lipid signalling to better understand the mechanisms by which they contribute to disease pathology.
- RNA-targeting compounds. Most drugs exert their principal biological effects by binding to a protein target. However, it is becoming increasingly clear that many RNA molecules also play key roles in disease. Therefore, there is a keen interest in developing systematic approaches to design compounds that bind specifically to RNA targets to modify their function. We have demonstrated that our REFiL strategy can be applied successfully to identify compounds that bind RNA and are currently using this approach to develop RNA-binding compounds that inhibit viral replication.
In addition to the design and synthesis of new compounds, each of these programs involves a range of different techniques and methods including protein expression and purification, structural biology, biophysical binding assays as well as biochemical and biological evaluation of compound potency. Several of these projects also involve collaboration with industry partners.
Contact
Leave this here so that Accordion nested does not detect this CT as not existing. -
Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences
Project Description
Endogenous peptides and protein interfacial fragments can be exquisitely potent biomolecules. In this project we look at strategies to transform these often-overlooked leads into potent drugs. You will learn small molecule synthesis, peptide synthesis and fundamental in vitro drug development assays. There will also be opportunities to extend your skill set into areas including computational modelling, pharmacology, and in vivo experimentation.
Project supervisors
Nick Barlow, Phil Thompson, David Chalmers, Ray Norton.
Contact
Leave this here so that Accordion nested does not detect this CT as not existing. -
Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences
The Scammells research group utilise ligand and structure-based design approaches to identify novel bioactive compounds. A long-term emphasis of this work has been on the development of allosteric modulators and bitopic ligands acting at various therapeutically relevant G protein-coupled receptors (GPCRs). Current projects focus on the development of positive allosteric modulators acting at the A1 adenosine receptor (A1 AR)1 as well as the μ- and δ-opioid receptors,2 all of which have potential applications in the treatment of chronic pain and depression. Another project centres on the design, synthesis and pharmacological assessment of allosteric and bitopic ligands acting the M4 muscarinic acetylcholine receptor for the treatment of neurocognitive disorders.3
Chemical biology projects focus on the development of chemical probes, primarily irreversible and fluorescent ligands, to elucidate receptor pharmacology with a recent example being the development of highly selective, fluorescent A1AR antagonists to quantify ligand binding parameters and visualise specific receptor distribution patterns in living cells.4
 Panel A: This figure shows the positive allosteric modulator MIPS521 bound to the A1 adenosine receptor. Panel B: Live-cell confocal imaging studies using a fluorescent A1AR antagonist. Panel C: Shows the potent dual inhibitor, MIPS1778, bound to the M17 aminopeptidase enzyme.
Panel A: This figure shows the positive allosteric modulator MIPS521 bound to the A1 adenosine receptor. Panel B: Live-cell confocal imaging studies using a fluorescent A1AR antagonist. Panel C: Shows the potent dual inhibitor, MIPS1778, bound to the M17 aminopeptidase enzyme.The final project area relates to the development of aminopeptidase inhibitors as anti-malarial agents. Plasmodium parasites rely on these enzymes to digest hemoglobin and obtain essential amino acids needed for growth and survival. This project employs structure-based design approaches to identify potent dual inhibitors of two aminopeptidase enzymes, namely PfMAP M1 and M17.5
All projects involve the design and synthesis of new chemical entities and are conducted in conjunction with pharmacology, biochemistry and/or structural biology collaborators, which provides the opportunity to gain cross-disciplinary experience in these important areas.
Key publications
- Structural Basis of Action of an Allosteric Non-opioid Analgesic Drug, C.J. Draper-Joyce, … A. Glukhova*, W.L. Imlach*, A. Christopoulos*, Nature, 2021, 597, 571-576.
- The Design, Synthesis and Evaluation of Novel 9-Arylxanthenedione-based Allosteric Modulators for the δ-Opioid Receptor, O. Deo, … C. Valant*, P.J. Scammells*, J. Med. Chem. 2022, 65, 12376-12385.
- Discovery of 2-Methyl-5-(1H-pyrazol-4-yl)pyridines and Related Heterocycles as Promising M4 mAChR Positive Allosteric Modulators for the Treatment of Neurocognitive Disorders, B. Liu, … B. Capuano*, C. Valant*, P.J. Scammells*, J. Med. Chem. 2024, in press.
- Development and Application of Subtype-Selective Fluorescent Antagonists for the Study of the Human Adenosine A1 Receptor in Living Cells, E. Comeo, … M.L. Halls*,L.T. May*, P.J. Scammells*, J. Med. Chem.2021, 64, 6670-6695.
- Structure-based Development of Potent Plasmodium falciparum M1 and M17 Aminopeptidase Selective and Dual inhibitors via S1′-region Optimisation, P.P.S. Calic, …. S. McGowan*, P.J. Scammells*, Eur. J. Med. Chem.2023, 248, 115051.
Contact
Leave this here so that Accordion nested does not detect this CT as not existing.
-
Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences
Nicotinamide adenine dinucleotide (NAD+) is an essential cofactor and signalling metabolite required for cellular energy production, DNA repair, stress responses, and neuronal health. Growing evidence indicates that impaired NAD+ biosynthesis and excessive NAD+ consumption contribute to the onset and progression of numerous neurodegenerative disorders, including Alzheimer's disease, Parkinson's disease, and motor neuron disease. Consequently, restoring NAD+ homeostasis has emerged as a promising therapeutic strategy for combating neurodegeneration.
This program aims to discover and develop novel small-molecule ligands that modulate key enzymes controlling neuronal NAD⁺ metabolism. By targeting both NAD⁺ biosynthetic and degradative pathways, we seek to establish innovative therapeutic approaches that maintain neuronal resilience, prevent metabolic dysfunction, and delay neurodegeneration. Specifically, the program will:
- Discover and optimise novel ligands using fragment-based drug design. Using state-of-the-art fragment screening, biophysical assays, structural biology, and computational design approaches, we will identify fragment molecules that modulate enzymes involved in NAD+ biosynthesis, particularly nicotinamide mononucleotide adenylyltransferase (NMNAT). These studies will provide a foundation for the rational optimisation of fragment hits into potent lead compounds capable of enhancing neuronal NAD⁺ production and restoring metabolic function in neurodegenerative disease.
- Develop targeted protein degraders to reduce pathological NAD+ consumption. CD38 is a major NAD+-consuming enzyme whose expression and activity increase during ageing, neuroinflammation, and neurodegeneration, contributing to the depletion of intracellular NAD⁺ pools. This project will explore innovative targeted protein degradation approaches to selectively eliminate CD38 and restore NAD+ homeostasis. By leveraging emerging induced-proximity technologies and degrader design principles, we aim to establish new therapeutic modalities for reducing pathological NAD⁺ consumption and improving neuronal function.
- Develop substrate-coupled inhibitors against NAD+ consumption. Enzymes often recognise their natural substrates with exceptional affinity and specificity, which can be exploited to enhance the potency and drug-like properties of small-molecule inhibitors. This project will establish a ligand discovery platform for the development of substrate-coupled inhibitors targeting therapeutically important NAD+-consuming enzymes, including SARM1 and CD38. Using integrated biochemical and computational approaches, we will identify small-molecule ligands that undergo target-catalysed coupling with the substrate to generate highly potent inhibitors. Structure-guided optimisation will then be used to enhance their affinity, selectivity, and drug-like properties, delivering novel chemical probes and potential therapeutic leads for neurodegenerative disease.
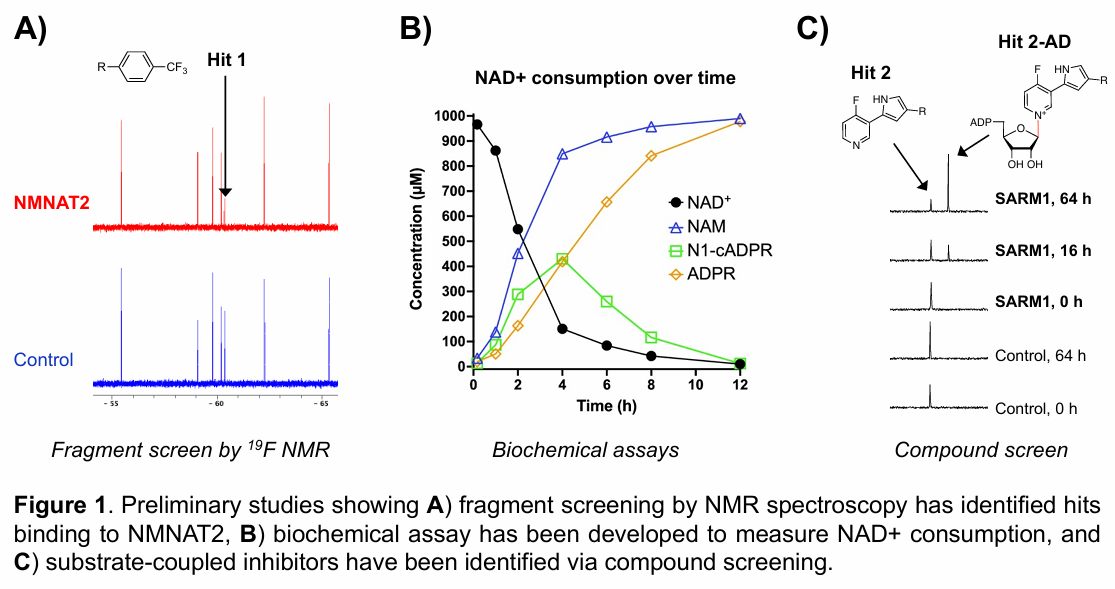
Preliminary work has established key assay platforms, structural models, and ligand discovery pipelines that enable the identification and optimisation of small molecules targeting NAD+ metabolic enzymes. These capabilities position the program to translate mechanistic insights into novel therapeutic candidates for neurodegenerative diseases.
Multiple Honours and PhD projects are available within this program, offering opportunities to work across fragment-based lead generation, targeted protein degradation, and mechanism-based ligand design. Students will receive interdisciplinary training in chemical biology, biochemistry, biophysics, structural biology, computational design, and medicinal chemistry, while contributing to the development of next-generation therapeutics for neurodegenerative diseases.
Contact
Leave this here so that Accordion nested does not detect this CT as not existing. -
Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences
Metabolites are not only the fundamental building blocks of life but also serve as critical signalling molecules that regulate a wide range of biological processes. Many of these functions are mediated through direct interactions with proteins, whereby metabolites modulate protein activity, stability, and cellular function. Dysregulation of metabolite–protein interactions has been implicated in numerous human diseases, yet the metabolite–protein interactome remains largely unexplored, representing a significant and underutilised avenue for therapeutic discovery.
This program aims to identify, characterise, and re-engineer novel metabolite-protein interactions for drug design, with a particular focus on metabolites and proteins involved in nicotinamide adenine dinucleotide (NAD+) metabolism. NAD+ is a central metabolic and signalling hub whose dysregulation is linked to neurodegenerative, inflammatory, and infectious diseases. By uncovering new mechanisms through which metabolites regulate protein function, this program seeks to establish innovative strategies for therapeutic intervention. Specifically, the program will:
- Systematically identify metabolite–protein interactions using an integrated discovery pipeline that combines state-of-the-art biophysical, chemical biology, and computational approaches.
- Characterise validated interactions at the molecular level through biochemical, biophysical, and structural studies to elucidate their mechanisms and biological significance.
- Design and develop small-molecule modulators capable of mimicking, disrupting, or reprogramming metabolite–protein interactions to achieve therapeutic benefit.
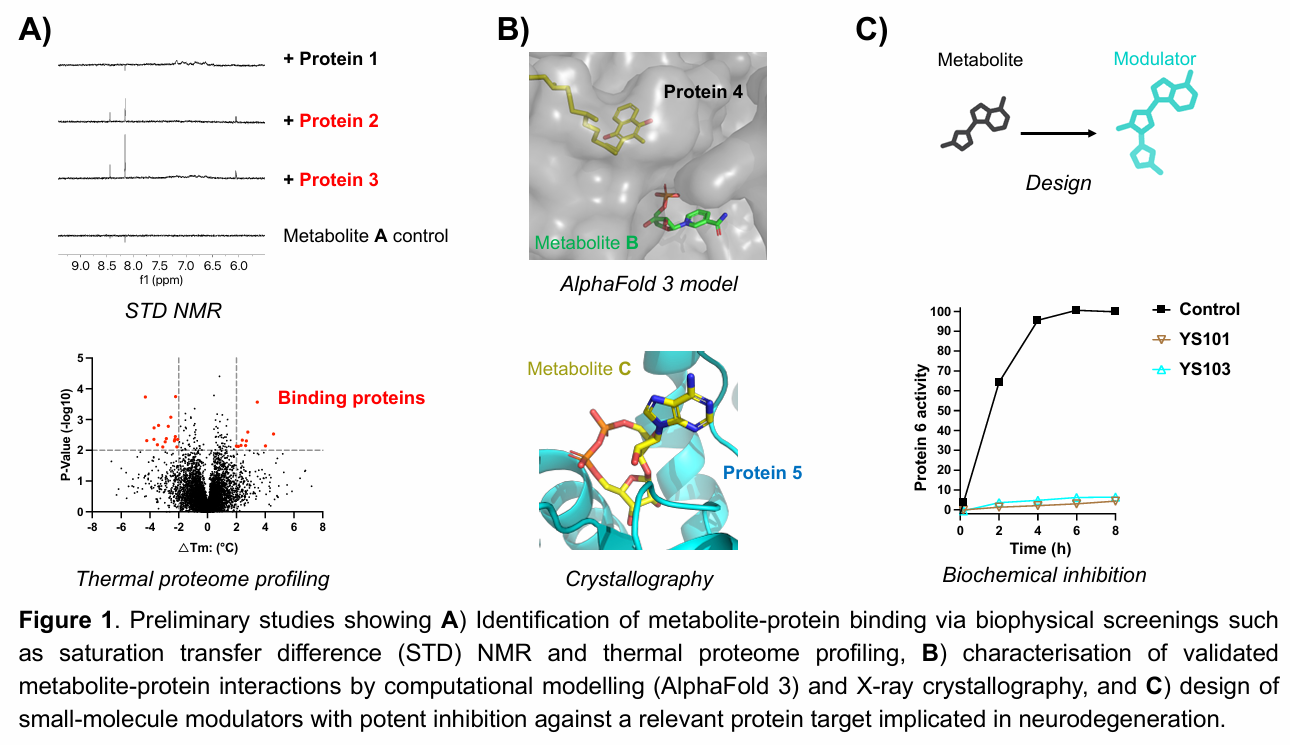
Preliminary studies have demonstrated the feasibility of this approach, leading to the discovery and molecular characterisation of previously unrecognised metabolite–protein interactions and the development of strategies to modulate them for therapeutic applications (Figure 1).
Multiple Honours and PhD projects are available within this program, offering opportunities to investigate distinct metabolites and protein targets involved in neurodegenerative, inflammatory, and infectious diseases. Students will gain interdisciplinary training spanning chemical biology, biochemistry, computational modelling, and drug discovery.
Contact
Leave this here so that Accordion nested does not detect this CT as not existing.